



La enfermedad de Whipple (EW) es una enfermedad infecciosa multisistémica producida por la bacteria Tropheryma whipplei (TW). Inicialmente fue descrita en 1907 por George H. Whipple como una «lipodistrofia intestinal», en un paciente con pérdida ponderal, poliartritis, diarrea, malabsorción y adenopatías mesentéricas1. La sintomatología neurológica en ausencia de clínica sistémica es infrecuente2.
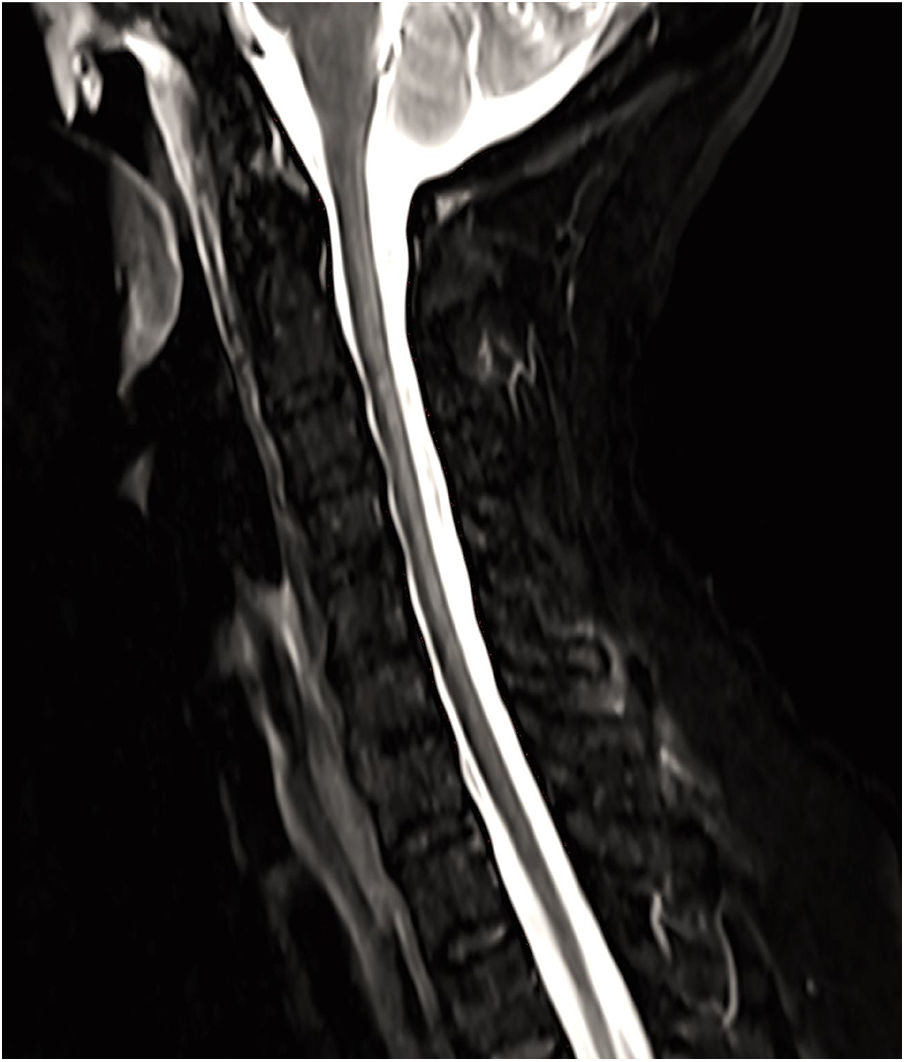
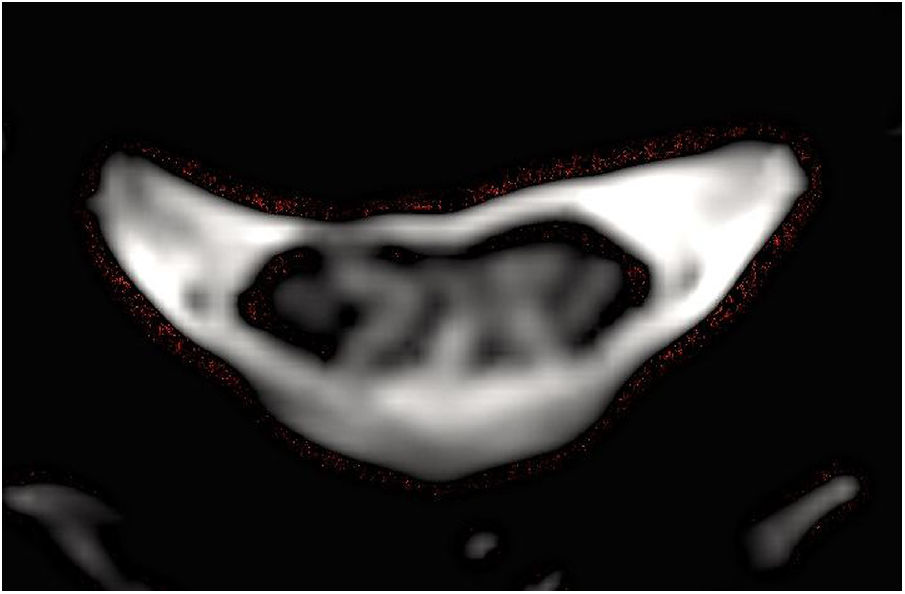
Presentamos un caso de EW que se manifiesta con clínica medular de manera aislada. Se trata de una mujer de 66 años que consulta por la alteración de la marcha de un año de evolución sin clínica a otro nivel, la exploración mostró disminución de sensibilidad profunda en miembros inferiores, sin datos de piramidalismo, siendo el resto del examen neurológico y sistémico normal. Los potenciales evocados somatosensoriales señalaron un defecto en la vía somestésica cordonal posterior, y con el fin de descartar una mielopatía, se llevó a cabo una resonancia magnética cervical sin contraste, que detectó una lesión hiperintensa en T2 comprometiendo los cordones posteriores desde C2 hasta T1, sin edema asociado (figs. 1 y 2), siendo el parénquima cerebral normal. Con el propósito de encontrar la etiología de dicha lesión, se llevó a cabo un análisis en sangre incluyendo bioquímica completa y estudio inmunológico, que no mostraron alteraciones de significación sugerentes de enfermedades autoinmunes sistémicas, metabólicas, carenciales o de otra índole. Puesto que en el diagnóstico diferencial se incluyen enfermedades infecciosas víricas y bacterianas, se completó la valoración con análisis microbiológico en sangre, determinando la presencia de PCR positiva para TW. Por esta razón, se llevó a cabo una punción lumbar realizando en LCR un análisis bioquímico, microbiológico, inmunológico y citológico, todo ello con resultados normales, a excepción de PCR positiva para TW, al igual que en determinación sanguínea. Se trató de aislar el material genético de dicho microorganismo en heces y saliva, con resultado negativo. Se detectó nuevamente PCR positiva para TW en una segunda determinación en sangre realizada 15 días más tarde. Así, se inició tratamiento con ceftriaxona intravenosa durante 14 días con buena respuesta clínica y continuando de forma prolongada con trimetoprim-sulfametoxazol vía oral, produciéndose clara mejoría en la marcha, con estabilidad de la lesión medular en estudio de resonancia magnética realizado meses más tarde. Asimismo, se produjo una negativización de la PCR para TW en sangre y líquido cefalorraquídeo.


La EW es una enfermedad infecciosa con manifestación multisistémica, aunque destaca la clínica digestiva y articular. Aunque la incidencia exacta no es del todo conocida3, el compromiso del sistema nervioso central es frecuente, apareciendo en un 43% de casos4, aunque no como presentación aislada, que se produce únicamente en el 5% de los pacientes5.
Los síntomas neurológicos son inespecíficos, y su presentación variada, desde alteración del nivel de consciencia, deterioro cognitivo, crisis epilépticas, mioclonías, compromiso cerebeloso, hipotalámico, neuropatías craneales y periféricas, y trastornos extrapiramidales5,6. La miorritmia oculomasticatoria y la miorritmia oculofacial-esquelética son muy características de esta entidad, y se consideran signos patognomónicos7,8.
Si bien existen casos de mielopatía asociada a otras lesiones inflamatorias en diferentes puntos del sistema nervioso central, como el cerebro, quiasma óptico y fosa posterior, caso del paciente descrito por Kremer et al.9, mucho menos frecuente aún es su presentación en forma de mielopatía aislada. El primer caso fue descrito por Clarke et al. en 1998 en una paciente de 62 años10, posteriormente Messori y Salvolini11 y Messori et al.12 describieron el caso de una paciente de 65 años con una lesión medular cérvico-torácica y curso clínico remitente-recurrente atribuida a infección por TW, desarrollando en fases posteriores lesiones cerebrales. En 2005, Schröter et al.13 aportaron el caso de un paciente de 50 años con una lesión cervical desde la unión bulbomedular hasta la región dorsal superior, sin compromiso cerebral, con buena respuesta a la terapia antibiótica frente a TW, detectado mediante PCR en sangre. En nuestro conocimiento, la paciente que describimos se trata del cuarto caso conocido, el primero en nuestro país, en el cual la EW se manifiesta como una mielopatía aislada, en forma de un síndrome cordonal posterior. Los casos descritos guardan ciertas similitudes como la edad de presentación (entre la sexta y séptima décadas de la vida), los síntomas de mielitis crónica progresiva, el compromiso cervical extenso en forma de lesión hiperintensa, así como la clara mejoría con terapia antibiótica.
El diagnóstico diferencial debe llevarse a cabo con otras enfermedades infecciosas bacterianas, víricas y parasitarias, con enfermedades desmielinizantes como la esclerosis múltiple, la neuromielitis óptica o la encefalomielitis aguda diseminada. También debe excluirse etiología vascular (isquemia o hemorragia), enfermedad tumoral o paraneoplásica, así como enfermedades sistémicas (lupus eritematoso sistémico, enfermedad de Behçet, síndrome de Sjögren, sarcoidosis) o enfermedades carenciales en forma de deficiencias vitamínicas.
La existencia de pocos casos en la literatura, así como el carácter inespecífico de los síntomas pueden dificultar el diagnóstico y, por tanto, retrasar el inicio del tratamiento. Debemos tener presente que la clínica neurológica puede ser la primera manifestación de la EW, pudiendo aparecer posteriormente otros síntomas más característicos14. En cualquier caso, la falta de confirmación microbiológica no debe retrasar el inicio del tratamiento en caso de sospecha clínica de EW. Igualmente se recomienda realizar tratamiento de prueba con antibioterapia dirigida antes de realizar una biopsia medular.
En conclusión, la sintomatología medular es una complicación muy infrecuente de la infección por TW, de modo que debemos tener en cuenta la posibilidad de una infección por dicho agente etiológico en casos de mielopatía aislada de etiología no aclarada, incluso en ausencia de síntomas sistémicos, lo cual requiere un alto nivel de sospecha clínica. La importancia del diagnóstico precoz reside en la existencia de un tratamiento efectivo que puede eliminar la sintomatología e incluso evitar la progresión de la enfermedad hacia otros órganos o regiones del sistema nervioso, siendo una enfermedad potencialmente mortal en caso de no recibir tratamiento dirigido.
FinanciaciónLa presente investigación no ha recibido ninguna beca específica de agencias de los sectores público, comercial, o sin ánimo de lucro.

