




La hipertensión arterial pulmonar se encuentra comúnmente en adultos con cardiopatías congénitas. De acuerdo con el tipo de defecto, el momento de la corrección y la repercusión hemodinámica será la magnitud del compromiso y a su vez, un determinante esencial en la posibilidad de realizar manejo correctivo en aquellos pacientes diagnosticados de manera tardía. Se hizo una revisión de la información disponible en cuanto a la clasificación, el diagnóstico y el manejo de acuerdo con la posibilidad de intervención, el tratamiento general y el uso de vasodilatadores pulmonares, con énfasis en las recomendaciones especiales para el manejo de los pacientes con síndrome de Eisenmenger.
Pulmonary arterial hypertension is commonly found in adults with congenital heart diseases. The magnitude of the compromise will depend on the type of defect, the time of the correction, and the haemodynamic repercussion, and, in turn will be an essential determining factor in the possibility of performing corrective management in those patients with delayed diagnoses. A review is presented on the information available as regards the classification, diagnosis, and management depending on the possibility of intervention, the general treatment, and use of pulmonary vasodilators, with an emphasis on the special recommendations for the management of patients with Eisenmenger syndrome.
La hipertensión arterial pulmonar asociada a la enfermedad cardiaca congénita, es un tipo de hipertensión arterial pulmonar con características y pronóstico diferentes a la debida a otras etiologías, que requiere manejo en centros con experiencia en el tratamiento de pacientes adultos con cardiopatías congénitas e hipertensión pulmonar.
Todos los defectos congénitos cardiacos en los cuales existen comunicaciones grandes intra- o extracardiacas, llevan a una sobrecarga de presión y volumen de la circulación pulmonar, lo cual favorece el desarrollo de hipertensión arterial pulmonar, con menos probabilidad de ocurrir cuando la corrección se realiza de manera temprana en la infancia1,2.
En la actualidad se observa un crecimiento continuo de la población de pacientes adultos con cardiopatías congénitas, a una tasa aproximada del 5% por año3. En Estados Unidos de calcula que hay más de un millón de adultos con cardiopatías congénitas, con un porcentaje estimado del 10% con hipertensión arterial pulmonar, alcanzando un 30% entre aquellos con defectos no corregidos4, de los cuales un 50% progresa a síndrome de Eisenmenger, una condición que se traduce en cortocircuito de derecha a izquierda, cianosis y compromiso multisistémico5. En los registros de hipertensión pulmonar del adulto del Reino Unido de 2012, la hipertensión arterial pulmonar asociada a enfermedad cardiaca congénita fue responsable del 30,2% de los casos de hipertensión arterial pulmonar, cifra similar a la debida a las formas idiopáticas de hipertensión arterial pulmonar (33,6%) y un poco mayor que la de hipertensión arterial pulmonar asociada a enfermedades del tejido conectivo (28,3%)2.
Aunque a la fecha en Colombia no se cuenta con datos estadísticos de pacientes cardiópatas congénitos adultos, dadas las condiciones geográficas, socioeconómicas y culturales puede presumirse que la población no corregida y por tanto el síndrome de Eisenmerger, representan un número importante entre los pacientes con hipertensión arterial pulmonar asociada a enfermedad cardiaca congénita. Por consiguiente, es indispensable identificar y tratar en forma adecuada a este grupo de pacientes para mejorar su calidad de vida y reducir la morbilidad y mortalidad, considerando manejo correctivo cuando aún sea posible.
ClasificaciónLos pacientes con hipertensión arterial pulmonar asociada a enfermedad cardiaca congénita, se pueden clasificar en cuatro grupos6,7 (tabla 1).
Fenotipos y características clínicas de la hipertensión arterial pulmonar asociada a enfermedad cardiaca congénita
| Grupo 1: síndrome de Eisenmenger |
| Defectos grandes intra- o extracardiacos |
| Cortocircuito invertido o bidireccional |
| Aumento severo de la RVP |
| Cianosis, eritrocitosis y compromiso multiorgánico |
| Grupo 2: cortocircuitos sistémico-pulmonares |
| Defectos moderados a grandes |
| Incremento leve a moderado de la RVP |
| Cortocircuito de izquierda a derecha |
| Cianosis no presente en reposo |
| Grupo 3: hipertensión arterial pulmonar coincidencial/asociada a pequeños defectos |
| Marcado aumento de la RVP en presencia de pequeños defectos cardiacos (DSA <2 cm y DSV <1 cm) |
| Defectos que no son responsables de la RVP elevada |
| Cuadro clínico similar a hipertensión arterial pulmonar idiopática |
| Grupo 4: hipertensión arterial pulmonar luego de cirugía cardiaca correctiva |
| Cardiopatía congénita reparada |
| Hipertensión pulmonar que persiste luego de cirugía |
| Hipertensión pulmonar que reaparece o se desarrolla meses a años luego de la cirugía |
| No hay defectos residuales significativos o secuelas de cirugías previas |
Modificado de Simonneau, et al6,7. RVP: resistencia vascular pulmonar, DSA: defecto septal atrial, DSV: defecto septal ventricular, HAP: hipertensión arterial pulmonar, HAPi: hipertensión arterial pulmonar idiopática.
La causa más común de hipertensión arterial pulmonar asociada a enfermedad cardiaca congénita son los defectos no corregidos con cortocircuitos sistémico-pulmonares ventriculares o de grandes arterias8,9, en los que existe un incremento en el flujo sanguíneo pulmonar con un nivel sistémico de presión que lleva a cambios proliferativos en la arquitectura pulmonar y produce un incremento severo en la resistencia vascular pulmonar (RVP) que con frecuencia finaliza en inversión del cortocircuito (síndrome de Eisenmerger)1. En 1958, Wood describió el síndrome de Eisenmerger como una hipertensión arterial pulmonar con presión de la arteria pulmonar a niveles sistémicos atribuible a RVP mayor a 10 unidades Wood (UW) y consecuente cortocircuito invertido o bidireccional ventricular o de las grandes arterias, lo cual refirió como cortocircuitos centrales1.
El factor etiológico más importante para presentar hipertensión arterial pulmonar asociada a enfermedad cardiaca congénita es la presión más que el flujo, de ahí que los cortocircuitos sistémico-pulmonares grandes pretricuspídeos, sean típicamente lesiones de baja presión, que con menor probabilidad se traducirán en hipertensión arterial pulmonar. Cuando estas lesiones terminan en hipertensión arterial pulmonar, ésta generalmente ocurre de manera tardía en la vida10. Según datos de los registros CONCOR y Danés, el riesgo de desarrollar hipertensión arterial pulmonar es del 7% en los defectos septales atriales (DSA), del 11% en los defectos septales ventriculares (DSV) y del 41% en los defectos septales atrioventriculares (DSAV). Los pacientes con DSV tienen riesgo dos veces mayor de presentar síndrome de Eisenmerger que quienes tienen DSA8, pero aquellos con lesiones pretricuspídeas que desarrollan síndrome de Eisenmerger tienen peor pronóstico y malos resultados, hecho que fue confirmado en el registro español REHAP, en el que se encontró una mortalidad 2,6 veces mayor en quienes tenían lesiones pretricuspídeas (DSA) en comparación con aquellos con cortocircuitos postricuspídeos, presentando un fenotipo más agresivo que el síndrome de Eisenmerger, con una tasa de deterioro más rápida y un panorama peor, simulando una hipertensión arterial pulmonar idiopática (HAPi)11–13.
En los pacientes con cortocircuitos postricúspides la hipertensión arterial pulmonar está presente desde antes del nacimiento, de modo que el ventrículo derecho está “acostumbrado” a una poscarga elevada. En contraste, en los pacientes con cortocircuitos atriales, el ventrículo derecho se vuelve una bomba de volumen de pared delgada al nacimiento, hasta que de manera tardía en la vida hay un incremento en la RVP que lo desafía a volverse una bomba de alta presión14. En tales casos el ventrículo derecho está mal adaptado, comportándose como el ventrículo derecho de los pacientes con hipertensión arterial pulmonar idiopática, con dilatación temprana y falla11.
Otros subtipos de cardiopatías congénitas asociadas con un riesgo incrementado de desarrollar hipertensión arterial pulmonar incluyen defectos cardiacos reparados y defectos pequeños restrictivos teóricamente no significativos desde el punto de vista hemodinámico. Estos defectos pequeños en presencia de hipertensión arterial pulmonar se consideran lesiones coincidentes. En este tipo la hipertensión arterial pulmonar es usualmente severa y puede comportarse similar a la hipertensión arterial pulmonar idiopática en cuanto a fisiología, respuesta al tratamiento y resultados a largo plazo15.
Cuando se realiza el cierre tardío de los cortocircuitos, en especial cuando son lesiones posestenóticas, los pacientes pueden continuar con hipertensión arterial pulmonar a pesar de la corrección exitosa, como ocurre en el caso de DSV, ventana aortopulmonar y ductus arterioso persistente (DAP), o puede desarrollarse de manera tardía en la vida, aun cuando la intervención de la lesión se realizó en la infancia y en ausencia de defectos residuales hemodinámicamente significativos. Este grupo de pacientes con hipertensión arterial pulmonar asociada a enfermedad cardiaca congénita postquirúrgica, tienen tasas de supervivencia menores que su contraparte de pacientes con síndrome de Eisenmerger, con el pronóstico de todos los tipos de hipertensión arterial pulmonar asociada a enfermedad cardiaca congénita11,13.
Un grupo particular son los pacientes llevados a procedimientos de switch atrial paliativo (reparación de Mustard o Senning) y/o cirugías correctivas en la infancia para D-transposición de grandes arterias (DTGA), los cuales parecen tener un riesgo aumentado de desarrollar hipertensión arterial pulmonar, sin una causa clara, ya que la mayoría de estas reparaciones se lleva a cabo en las primeras semanas de vida. Es posible que la enfermedad vascular pulmonar esté presente en un subgrupo de pacientes antes del reparo quirúrgico, lo cual puede ocurrir hasta en un 10% de los casos16,17.
No es raro que el paciente con hipertensión arterial pulmonar asociada a enfermedad cardiaca congénita con malformaciones “ocultas” como DAP, defectos tipo seno venoso, drenaje venoso anómalo parcial o ventanas aortopulmonares, sea mal diagnosticado como hipertensión arterial pulmonar idiopática2.
La circulación tipo Fontan tiene una consideración fisiológica importante. Esta alteración anatómica, independiente de la reconstrucción quirúrgica, se basa en un flujo venoso sistémico pasivo entregado a baja presión (no hay una bomba subventricular pulmonar) para suplir adecuadamente la circulación pulmonar. En estos pacientes, un incremento marginal en la presión arterial pulmonar o cambios adversos en la RVP pueden alterar dramáticamente el circuito y llevar a una cascada de secuelas clínicas desfavorables2. Pese a las secuelas potenciales, los criterios para hipertensión pulmonar/hipertensión arterial pulmonar en estos pacientes comúnmente no se basan en la definición hemodinámica clásica, ya que la presión arterial pulmonar no puede ser muy alta14. Estos pacientes pueden tener lechos vasculares pulmonares anormales, así como respuesta subóptima a las terapias vasculares pulmonares, y requieren manejo agresivo para mejorar la descompensación clínica14.
Mientras la enfermedad vascular pulmonar en hipertensión arterial pulmonar asociada a enfermedad cardiaca congénita no difiere en cuanto a los hallazgos histológicos comparados con la hipertensión arterial pulmonar idiopática u otras formas de hipertensión arterial pulmonar, hay diferencias importantes en lo referente a la fisiopatología y al manejo2.
Aunque en el síndrome de Eisenmerger usualmente se presenta un compromiso multisistémico y hay menor expectativa de vida que en los pacientes con cardiopatías congénitas sin hipertensión arterial pulmonar, su pronóstico es mejor en comparación con los pacientes con hipertensión arterial pulmonar idiopática, tienen mejores tasas de supervivencia, que alcanzan hasta la cuarta a quinta década de la vida y, en muchos casos, mejor calidad de vida2.
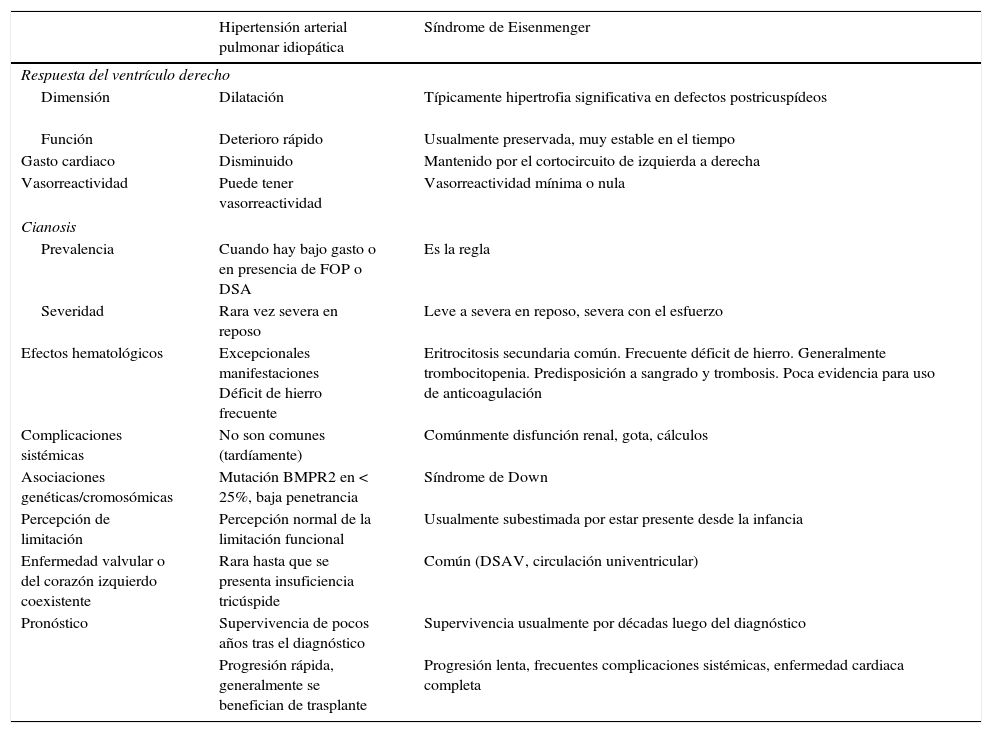
Síndrome de EisenmengerLa característica más relevante del síndrome de Eisenmerger, que lo diferencia de otros tipos de hipertensión arterial pulmonar, es la presencia de cianosis severa de larga duración, con sus efectos sistémicos subsecuentes y complicaciones potenciales (tabla 2).
Diferencias entre síndrome Eisenmenger e hipertensión arterial pulmonar idiopática
| Hipertensión arterial pulmonar idiopática | Síndrome de Eisenmenger | |
|---|---|---|
| Respuesta del ventrículo derecho | ||
| Dimensión | Dilatación | Típicamente hipertrofia significativa en defectos postricuspídeos |
| Función | Deterioro rápido | Usualmente preservada, muy estable en el tiempo |
| Gasto cardiaco | Disminuido | Mantenido por el cortocircuito de izquierda a derecha |
| Vasorreactividad | Puede tener vasorreactividad | Vasorreactividad mínima o nula |
| Cianosis | ||
| Prevalencia | Cuando hay bajo gasto o en presencia de FOP o DSA | Es la regla |
| Severidad | Rara vez severa en reposo | Leve a severa en reposo, severa con el esfuerzo |
| Efectos hematológicos | Excepcionales manifestaciones Déficit de hierro frecuente | Eritrocitosis secundaria común. Frecuente déficit de hierro. Generalmente trombocitopenia. Predisposición a sangrado y trombosis. Poca evidencia para uso de anticoagulación |
| Complicaciones sistémicas | No son comunes (tardíamente) | Comúnmente disfunción renal, gota, cálculos |
| Asociaciones genéticas/cromosómicas | Mutación BMPR2 en < 25%, baja penetrancia | Síndrome de Down |
| Percepción de limitación | Percepción normal de la limitación funcional | Usualmente subestimada por estar presente desde la infancia |
| Enfermedad valvular o del corazón izquierdo coexistente | Rara hasta que se presenta insuficiencia tricúspide | Común (DSAV, circulación univentricular) |
| Pronóstico | Supervivencia de pocos años tras el diagnóstico | Supervivencia usualmente por décadas luego del diagnóstico |
| Progresión rápida, generalmente se benefician de trasplante | Progresión lenta, frecuentes complicaciones sistémicas, enfermedad cardiaca completa | |
Modificada de Dimopoulus, et al.2. FOP: foramen ovale permeable.
La cianosis en el síndrome de Eisenmerger es el reflejo del cortocircuito de derecha a izquierda, que ocurre debido a la elevación de la RVP y al aumento de la presión ventricular derecha, aunque también ayuda a mantener el gasto cardiaco con el ejercicio18. La cianosis conlleva cambios hematológicos significativos, incluyendo eritrocitosis secundaria, trombocitopenia y en ocasiones leucopenia19. La eritrocitosis secundaria es un mecanismo compensatorio cuyo objetivo es aumentar la capacidad de transporte de oxígeno para maximizar la entrega de oxígeno a los tejidos dada la cianosis2.
La cianosis crónica en el síndrome de Eisenmerger también lleva a anormalidades significativas en la coagulación, lo cual puede tener implicaciones clínicas. Existe riesgo aumentado de trombosis dentro de las arterias pulmonares centrales. Los pacientes con síndrome de Eisenmerger tienen al mismo tiempo, mayor riesgo de sangrado, como epistaxis, menorragia y hemoptisis, lo cual generalmente es autolimitado, pero en ocasiones puede ser amenazante para la vida2. Por lo anterior, en la actualidad no hay un consenso en cuanto a la anticoagulación rutinaria, aunque muchos pacientes pueden requerirla por otras indicaciones (embolia pulmonar, arritmias, etc.). No es infrecuente la presencia de hiperuricemia, con crisis gotosa asociada y riesgo incrementado de litiasis, arritmias y absceso cerebral. En estos pacientes se requiere el uso de profilaxis para endocarditis bacteriana y vacunación contra neumococo e influenza5,14.
DiagnósticoEl diagnóstico de enfermedad vascular pulmonar requiere especial atención y experticia en hipertensión arterial pulmonar-enfermedad cardiaca congénita. En pacientes con grandes cortocircuitos postricúspides, el diagnóstico de síndrome de Eisenmerger puede confirmarse sólo con ecocardiografía: la cianosis en reposo o inducida por el ejercicio, con cortocircuito bidireccional de baja velocidad a través de un DSV grande, en ausencia de estenosis pulmonar, puede ser suficiente para indicar la presencia de niveles sistémicos de presión arterial pulmonar2. Sin embargo, ante DSA o cortocircuitos de izquierda a derecha, es indispensable el cateterismo cardiaco para el diagnóstico de hipertensión arterial pulmonar a fin de guiar el manejo. Cuando existe un cortocircuito grande de izquierda a derecha, la estimación precisa de la RVP llega a ser esencial ya que el incremento en el flujo pulmonar por el cortocircuito puede causar un aumento significativo de la presión arterial pulmonar con poco o ningún aumento de la RVP20. Identificar hipertensión arterial pulmonar con base sólo en la presión arterial pulmonar media (PAPm) > 25mm Hg y presión normal en la aurícula derecha, no es suficiente para establecer el diagnóstico de enfermedad vascular pulmonar en cardiopatía congénita9. En pacientes con DSA por el aumento del flujo pulmonar puede encontrarse elevación de la presión arterial pulmonar, manteniendo RVP en rangos cercanos a lo normal, en cuyo caso se podrían considerar candidatos para cierre del defecto9,21. De manera inversa, los pacientes con flujo pulmonar muy bajo, incluyendo aquellos con circulación tipo Fontan (univentricular), pueden tener un aumento en la RVP mas no de la presión arterial pulmonar, y podrían beneficiarse de manejo para hipertensión arterial pulmonar2.
En paciente con fisiología univentricular, por las condiciones descritas del circuito, se considera hipertenso si existen RVP > 3 UW y un gradiente transpulmonar (GTP) mayor a 6. Estos datos son tomados de pacientes pediátricos debido a que el universo más grande de este tipo de fisiología se encuentra en este grupo poblacional22.
Frente a cortocircuitos es importante estimar el Qp:Qs, no sólo para calcular la RVP sino para cuantificar la magnitud de los mismos y así definir la posibilidad de corrección. Para este propósito, se usa el principio de Fick7. Nunca se deberían usar métodos por termodilución en pacientes con cortocircuitos intracardiacos. Finalmente, los estudios de vasorreactividad con vasodilatadores pulmonares como el óxido nítrico, deberían reservarse para los pacientes con hipertensión arterial pulmonar-enfermedad cardiaca congénita, en quienes se desea evaluar la posibilidad de corrección9,21,23. Aunque la vasorreactividad podría tener alguna implicación pronóstica en el grupo de pacientes con hipertensión arterial pulmonar-enfermedad cardiaca congénita, no hay datos del beneficio potencial de los antagonistas del calcio en aquellos con hipertensión arterial pulmonar-enfermedad cardiaca congénita, a diferencia del respaldo para su uso en HAPi7.
Estrategias de manejoLa aproximación terapéutica de la hipertensión arterial pulmonar-enfermedad cardiaca congénita depende de la historia médica y quirúrgica, el estado clínico y el perfil hemodinámico. El objetivo del manejo puede ser de soporte, tratamiento con vasodilatadores e intervenciones con catéter y/o quirúrgicas.
El progreso en las técnicas quirúrgicas e intervencionistas en las recientes décadas, facilita hoy en día la corrección con bajo riesgo. Las lesiones hemodinámicamente significativas requieren manejo oportuno ya que las reparaciones tardías pueden ser perjudiciales para el paciente, en especial a largo plazo. No hay duda de que en los pacientes con síndrome de Eisenmerger, nunca deben cerrarse los defectos ya que el cortocircuito actúa como válvula aliviadora para el ventrículo derecho y el lecho vascular pulmonar, y ayuda a mantener el gasto cardiaco a través del cortocircuito de derecha a izquierda5. El éxito aparente del cierre y los buenos resultados a corto plazo (paciente vivo y sin cianosis), de ninguna manera son indicativos de pronóstico favorable a largo plazo, pues la enfermedad vascular pulmonar y la disfunción del ventrículo derecho usualmente progresan en las semanas, meses y años luego del cierre del cortocircuito13.
Manejo generalHay una serie de medidas generales, esenciales para mejorar la calidad de vida y los resultados en los pacientes con hipertensión arterial pulmonar-enfermedad cardiaca congénita, principalmente evitando los errores y abandonando terapias viejas deletéreas. El mito que los pacientes con síndrome Eisenmenger tienen una alta predisposición a complicaciones catastróficas debido a su elevada hiperviscosidad sanguínea, es ahora un reto. A diferencia de la policitemia rubra vera, los síntomas de hiperviscosidad y las complicaciones embólicas son raras en el síndrome de Eisenmerger2. A estos pacientes se les recomienda mantener una hidratación adecuada, evitar la inmovilización prolongada y diagnosticar y tratar el déficit de hierro. Las flebotomías, con el objetivo de disminuir los niveles de hemoglobina y hematocrito dentro de rangos normales (para pacientes no cianóticos), promueven el déficit de hierro y parecen incrementar, más que reducir, el riesgo de eventos cerebrovasculares5,24. Los pacientes con cardiopatía congénita cianozante, deberían ser tamizados para déficit de hierro, la cual es común incluso en ausencia de flebotomías, y precisan el inicio de reposición del mismo. Las flebotomías se reservan para aquellos con síntomas severos secundarios a la hiperviscosidad, en ausencia de deshidratación o déficit de hierro, y deben realizarse sólo con prescripción de grupos especializados y con reposición adecuada de volumen. Todos los líquidos intravenosos (IV) y medicación IV deben administrarse cuidadosamente, con uso de filtros de aire para evitar la embolia paradójica de aire2,5.
La hipertensión arterial pulmonar afecta significativamente la capacidad de ejercicio y la calidad de vida. El ejercicio extenuante y los esfuerzos isométricos extremos debe desaconsejarse, pero se recomienda continuar la actividad física dentro de sus propias capacidades2,25. El uso de oxígeno continuo durante el día y la noche no está avalado por la evidencia y puede llevar a dependencia sicológica y desacondicionamiento físico por la limitación de la movilización; podría tener alguna utilidad el uso nocturno en aquellos pacientes con cortocircuito bidireccional2. Los vuelos comerciales no están contraindicados en los pacientes con síndrome de Eisenmerger y en la mayoría no se requiere el uso de oxígeno suplementario26.
El embarazo conlleva un riesgo excesivamente elevado de mortalidad materna y fetal (grupo IV de riesgo de la Organización Mundial de la Salud) y está contraindicado (mortalidad tres veces mayor)27; por tanto, se recomienda terminar la gestación en caso de presentarse. Se hace énfasis en la anticoncepción efectiva, incluso considerando doble método sobre todo en aquellas mujeres que reciben inhibidores de endotelina, especialmente bosentan, dada la interacción con los compuestos basados en progesterona. Se ha encontrado que los compuestos con estrógenos aumentan el riesgo de trombosis, por lo que se debería restringir su uso2.
Los procedimientos bajo anestesia general e incluso la sedación, conllevan alto riesgo de complicaciones en pacientes con síndrome de Eisenmerger, así que deberían evitarse en la medida de lo posible. Sin embargo, si son esenciales, se deberían practicar en cuarto nivel de atención, donde se disponga de anestesiólogo cardiovascular y personal entrenado en el manejo de la hipertensión arterial pulmonar-enfermedad cardiaca congénita.
Manejo específicoEn los pacientes con hipertensión arterial pulmonar luego de cirugías correctivas y aquellos con defectos pequeños con hipertensión arterial pulmonar coincidentes, no hay indicación para manejo intervencionista o quirúrgico. Los pacientes con estas entidades tienen un comportamiento fisiológico similar al de la hipertensión arterial pulmonar idiopática, de ahí que también los sea el tratamiento. Para confirmar la fisiología en aquellos pacientes con hipertensión arterial pulmonar secundaria a cortocircuitos de izquierda a derecha por defectos moderados a grandes, con secuelas de leve a moderada elevación de la RVP, se requiere un cateterismo cardiaco para evaluación hemodinámica y pruebas de vasorreactividad. Si es posible se debería intentar ocluir el defecto para evaluar las consecuencias hemodinámicas de un cierre potencial.
Con el resultado de la RVP se define el paso siguiente en el manejo. Si la RVP es menor de 2,3 UW o indexada (IRVP) < 4 UW/m2, es posible considerar el cierre del defecto. En caso de tener RVP >8 UW o IRVP > 4,6 UW/m2 no se debería, en principio, optar por el cierre del defecto. En aquellos con valores intermedios, se evalúa cada caso y se define el beneficio teórico, ya que los resultados han sido contradictorios y aquellos con hipertensión arterial pulmonar residual luego de la corrección tienen un pronóstico más pobre, incluso peor que aquellos manejados médicamente, de modo que tal vez no sean buenos candidatos para cierre13. Una opción actual para este grupo de pacientes con valores intermedios es el inicio de manejo farmacológico para hipertensión arterial pulmonar y el reparo del defecto si hay caída de la RPV a niveles aceptables para realizarlo; esta propuesta está sustentada por reportes de casos, generalmente en pacientes con DSA12 (tabla 3).
Los pacientes con DSA y disfunción ventricular izquierda sistólica o diastólica, deberían ser tratados con precaución, ya que el DSA descomprime la aurícula izquierda. En tales casos, la oclusión total del defecto puede llevar a aumento súbito en la presión venosa pulmonar y desarrollo de edema pulmonar. La oclusión con dispositivos fenestrados o no realizar el cierre, es la opción más recomendable. La evaluación de la presión real de la aurícula izquierda, haciendo oclusión durante el estudio hemodinámico, es una alternativa a considerar para definir el impacto real2.
Manejo con vasodilatadores pulmonaresSíndrome de EisenmengerLas terapias avanzadas de hipertensión arterial pulmonar hoy en día se usan de manera rutinaria en este tipo de pacientes con el objetivo de mejorar la capacidad de ejercicio y, por consiguiente, la calidad de vida. Sin embargo, hay varios estudios que buscan específicamente terapias dirigidas para pacientes con síndrome de Eisenmerger que han permitido recomendar estos tratamientos. Se ha demostrado eficacia con el uso de inhibidores de la fosfodiesterasa tipo 5, antagonistas del receptor de endotelina, análogos de la prostaciclina y agonistas del receptor IP.
Los resultados del BREATHE-5, primer estudio doble ciego, de asignación aleatoria, controlado con placebo en pacientes con síndrome de Eisenmerger por DSA o DSV, clase funcional III, demostró que el bosentan, un inhibidor de la endotelina, disminuía significativamente la RVP y mejoraba la capacidad de ejercicio a las 16 semanas, con extensión del beneficio vista hasta el año de seguimiento28. De igual forma, se han adelantado estudios con inhibidores de la fosfodiesterasa y tratamiento combinado con bosentan y sildenafil que demuestran beneficio hemodinámico y/o clínico29–31. El efecto benéfico de la terapia con vasodilatadores pulmonares parece mantenerse durante varios años, pese a que los datos iniciales indicaban pérdida de la eficacia luego del primer año31–33.
Así mismo, se ha reportado el uso de epoprostenol IV en pacientes con síndrome de Eisenmerger, con efectos hemodinámicos favorables y mejoría en la capacidad física generalmente a largo plazo, aunque las líneas centrales predisponen a embolia paradójica y sepsis34. De manera equivalente, hay pequeños estudios con iloprost inhalado que parecen tener beneficio en variables clínicas con su administración35.
En aquellos con síndrome de Eisenmerger en quienes hay falla en la respuesta al tratamiento y continúan deteriorándose, el trasplante pulmonar con corrección del defecto cardiaco o el trasplante combinado corazón-pulmón llega a ser la opción terapéutica final36,37.
El papel potencial de los dispositivos de asistencia ventricular y el corazón artificial total en pacientes con hipertensión arterial pulmonar- asociada a cardiopatías congénitas del adulto (HAP-CCA) está aún en investigación, pero se cuenta con resultados preliminares prometedores38.
Hipertensión arterial pulmonar en pacientes con cortocircuitos sistémico-pulmonaresEl uso de terapias avanzadas en este tipo de pacientes aún es controversial ya que dada la persistencia del cortocircuito de izquierda a derecha se uso lleva a un aumento del flujo sanguíneo pulmonar comparado con el sistémico, lo cual podría acelerar la progresión de la enfermedad vascular pulmonar. Sin embargo, se ha demostrado que con estas terapias hay mejoría de la capacidad de ejercicio13. Por lo anterior, no es posible en la actualidad hacer recomendaciones a favor o en contra de su empleo.
Hipertensión arterial pulmonar en pacientes con defectos pequeños y en pacientes luego de defectos reparadosEn pacientes con hipertensión arterial pulmonar y defectos pequeños (coincidental), la hipertensión pulmonar se describe comúnmente como hipertensión arterial pulmonar idiopática con defectos cardiacos coexistentes, dado que el defecto solo no puede explicar la magnitud de la hipertensión arterial pulmonar. Parece que en estos pacientes la presencia de DSA mejora la supervivencia ya que permite la descompresión del ventrículo derecho, aunque esto no se ha demostrado por completo. Esta es la base teórica para recomendar la atrioseptostomía en los casos con hipertensión arterial pulmonar severa7. En vista de que el pronóstico y los hallazgos fisiopatológicos son similares, se recomienda manejo con vasodilatadores, anticoagulación -si aplica- y evitar el cierre de los defectos.
Los pacientes con hipertensión arterial pulmonar con historia de defectos cardiacos previos reparados, sin lesiones hemodinámicamente significativas, fueron incluidos en los estudios de las terapias vasodilatadoras, conjuntamente con la hipertensión arterial pulmonar idiopática y la hipertensión arterial pulmonar asociada a enfermedades del tejido conectivo. Su pronóstico es similar a las formas de hipertensión arterial pulmonar idiopática, de ahí que el tratamiento recomendado también lo sea2,7.
Fisiología de Eisenmenger en paciente con corazón univentricularEl uso de terapias avanzadas en esta población de pacientes no ha sido valorado ampliamente con estudios grandes, de asignación aleatoria, ya que los pacientes complejos usualmente son excluidos dada la dificultad en estimar la RVP. En ausencia de estenosis pulmonar que proteja la circulación pulmonar, la enfermedad vascular pulmonar se desarrolla durante la infancia, limitando el flujo sanguíneo pulmonar. Un incremento en el flujo pulmonar podría ser bien recibido y, en teoría, podría disminuir la cianosis periférica y mejorar la entrega de oxígeno a los tejidos. Sin embargo, aumentos significativos en el flujo pueden llevar a una sobrecarga de volumen del ventrículo único, el cual soporta ambas circulaciones, sistémica y pulmonar, y puede precipitarse disfunción ventricular39. No existe, por tanto, evidencia para recomendar los vasodilatadores pulmonares, los cuales se usan solo en casos seleccionados, de manera empírica. Podría hacerse una valoración hemodinámica invasiva o con resonancia cardiaca del Qp y Qs para apoyar la decisión en cuanto a su uso40.
En lo que concierne a la circulación tipo Fontan, el incremento en la presión arterial pulmonar o RVP puede tener efectos deletéreos que llevan a fallo del Fontan, caracterizado por falla cardiaca congestiva, arritmias, ascitis, enteropatía perdedora de proteínas y muerte. Aunque hay varios estudios de asignación no aleatoria que han mostrado eficacia en el ejercicio y la capacidad funcional41,42, dos estudios de asignación aleatoria (uno en el que se evalúa bosentan y en el otro sildenafil), fallaron en demostrar eficacia43,44. Pese a los resultados mixtos, el TEMPO, un gran estudio de asignación aleatoria, doble ciego, controlado con placebo mostró mejoría en la capacidad y el tiempo de ejercicio y la clase funcional45.
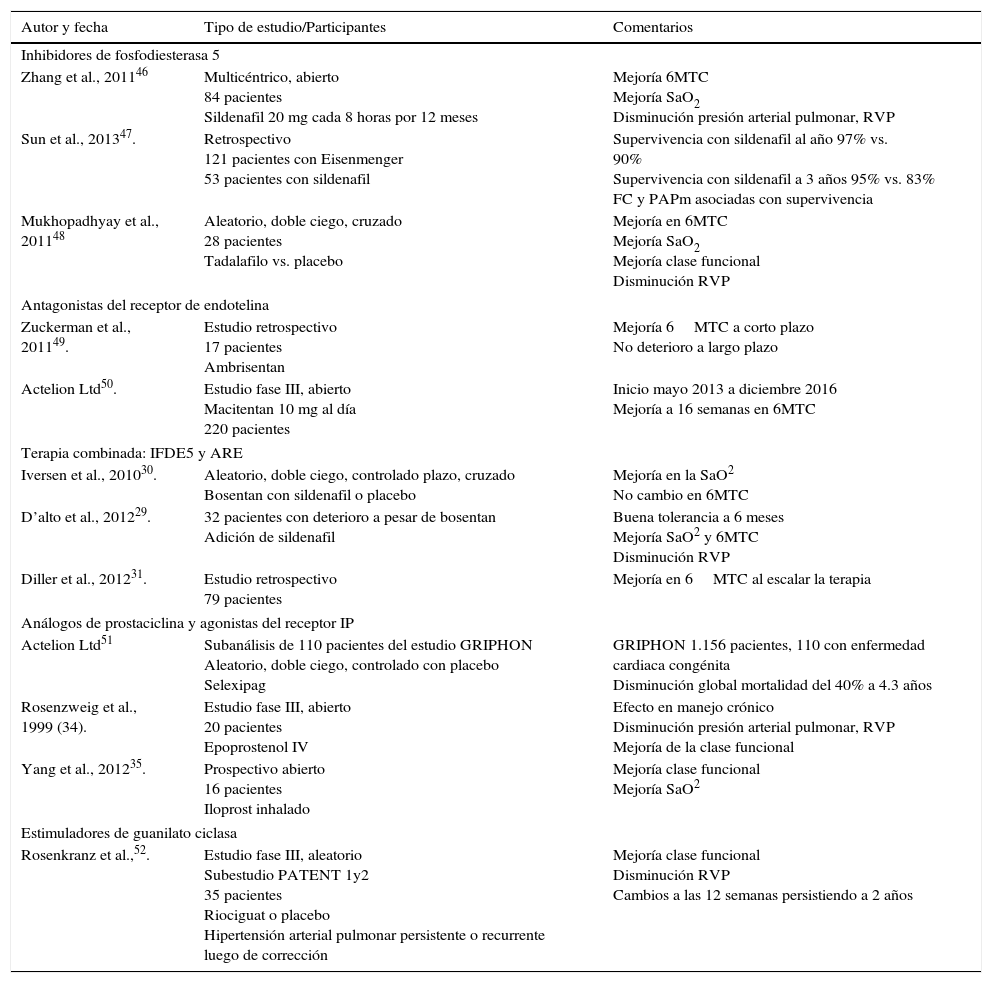
En la tabla 4 se resumen los vasodilatadores pulmonares con efecto benéfico en el tratamiento de la hipertensión arterial pulmonar-enfermedad cardiaca congénita.
Estudios con vasodilatadores pulmonares en pacientes hipertensión arterial pulmonar-enfermedad cardiaca congénita
| Autor y fecha | Tipo de estudio/Participantes | Comentarios |
|---|---|---|
| Inhibidores de fosfodiesterasa 5 | ||
| Zhang et al., 201146 | Multicéntrico, abierto 84 pacientes Sildenafil 20 mg cada 8 horas por 12 meses | Mejoría 6MTC Mejoría SaO2 Disminución presión arterial pulmonar, RVP |
| Sun et al., 201347. | Retrospectivo 121 pacientes con Eisenmenger 53 pacientes con sildenafil | Supervivencia con sildenafil al año 97% vs. 90% Supervivencia con sildenafil a 3 años 95% vs. 83% FC y PAPm asociadas con supervivencia |
| Mukhopadhyay et al., 201148 | Aleatorio, doble ciego, cruzado 28 pacientes Tadalafilo vs. placebo | Mejoría en 6MTC Mejoría SaO2 Mejoría clase funcional Disminución RVP |
| Antagonistas del receptor de endotelina | ||
| Zuckerman et al., 201149. | Estudio retrospectivo 17 pacientes Ambrisentan | Mejoría 6MTC a corto plazo No deterioro a largo plazo |
| Actelion Ltd50. | Estudio fase III, abierto Macitentan 10 mg al día 220 pacientes | Inicio mayo 2013 a diciembre 2016 Mejoría a 16 semanas en 6MTC |
| Terapia combinada: IFDE5 y ARE | ||
| Iversen et al., 201030. | Aleatorio, doble ciego, controlado plazo, cruzado Bosentan con sildenafil o placebo | Mejoría en la SaO2 No cambio en 6MTC |
| D’alto et al., 201229. | 32 pacientes con deterioro a pesar de bosentan Adición de sildenafil | Buena tolerancia a 6 meses Mejoría SaO2 y 6MTC Disminución RVP |
| Diller et al., 201231. | Estudio retrospectivo 79 pacientes | Mejoría en 6MTC al escalar la terapia |
| Análogos de prostaciclina y agonistas del receptor IP | ||
| Actelion Ltd51 | Subanálisis de 110 pacientes del estudio GRIPHON Aleatorio, doble ciego, controlado con placebo Selexipag | GRIPHON 1.156 pacientes, 110 con enfermedad cardiaca congénita Disminución global mortalidad del 40% a 4.3 años |
| Rosenzweig et al., 1999 (34). | Estudio fase III, abierto 20 pacientes Epoprostenol IV | Efecto en manejo crónico Disminución presión arterial pulmonar, RVP Mejoría de la clase funcional |
| Yang et al., 201235. | Prospectivo abierto 16 pacientes Iloprost inhalado | Mejoría clase funcional Mejoría SaO2 |
| Estimuladores de guanilato ciclasa | ||
| Rosenkranz et al.,52. | Estudio fase III, aleatorio Subestudio PATENT 1y2 35 pacientes Riociguat o placebo Hipertensión arterial pulmonar persistente o recurrente luego de corrección | Mejoría clase funcional Disminución RVP Cambios a las 12 semanas persistiendo a 2 años |
La prevalencia de las cardiopatías congénitas en los pacientes adultos continúa incrementándose debido a que se ha logrado una mayor supervivencia de aquellos intervenidos previamente gracias al aumento de la tecnología disponible y a la mayor disponibilidad de las herramientas diagnósticas, lo que a su vez se refleja en el número de pacientes que presentan o eventualmente desarrollarán hipertensión arterial pulmonar, y lleva a un importante deterioro de su calidad de vida y pronóstico a largo plazo. En la actualidad hay mayor disponibilidad de tratamientos que permiten impactar en forma positiva los desenlaces clínicos y hemodinámicos; por supuesto, estos deben realizarse en centros que dispongan de manejo especializado y con experiencia en el tratamiento de la hipertensión arterial pulmonar-enfermedad cardiaca congénita, ya que algunos pacientes seleccionados con defectos no corregidos podrían aún beneficiarse de manejo intervencionista y en aquellos en quienes no hay respuesta a la terapia y continúa el deterioro clínico, podrían ser considerados para terapias avanzadas como el trasplante.
Conflicto de interesesNinguno.