





El manejo farmacológico de la hipertensión arterial pulmonar se basa en agentes que actúan en tres vías principales: endotelina 1, prostaglandina I2 y óxido nítrico. La mayoría de estudios clínicos para aprobación de medicamentos desarrollados para tratar esta condición, han sido cortos y enfocados en el cambio en la caminata de 6 minutos. Al tener en cuenta que las diferentes formas de hipertensión arterial pulmonar tienen como denominador común para las alteraciones moleculares y celulares el entrecruzamiento celular con la pared vascular asociado a procesos inflamatorios e inmunes inapropiados, disbalance entre la síntesis y degradación de matriz extracelular, alteraciones genéticas (gen BMPR2 en hipertensión arterial pulmonar hereditaria) y epigenéticas, se requiere un mejor entendimiento de la fisiopatología de la enfermedad, lo cual permitirá desarrollar nuevos tratamientos o intervenciones en estos pacientes en cada uno de estos niveles.
The pharmacological management of pulmonary arterial hypertension is based on drugs that act on three main pathways: endothelin 1, prostaglandin I2, and nitric oxide. The majority of clinical studies for the approval of drugs developed to treat this condition have been short and focused on changes in the 6-minute walk test. On taking into account that the different forms of pulmonary arterial hypertension have cell cross-over with the vascular wall as a common denominator for the molecular and cellular changes associated with inappropriate inflammatory and immune processes, imbalance between synthesis and extracellular matrix degradation, genetic (BMPR2 gene in hereditary pulmonary arterial hypertension) and epigenetic alterations, a better understanding of the pathophysiology of the disease is required, which will help in the development of new treatments or interventions in these patients in each one of these levels.
Los tratamientos farmacológicos específicos aprobados para pacientes con hipertensión arterial pulmonar incluyen agentes que tienen efectos vasoactivos y de modulación en tres vías principales: endotelina 1 (ET-1), prostaglandina I2 (PGI-2) y óxido nítrico (ON)1.
Los medicamentos para el tratamiento de la hipertensión arterial pulmonar, a excepción de macitentan y selexipag, fueron aprobados con base en estudios clínicos de corta duración (12-24 semanas), en donde se enfocaron principalmente en la capacidad de ejercicio evaluada mediante el cambio en la caminata de 6 minutos. La mayoría de estos estudios demostró un retraso en el tiempo a empeoramiento clínico (TAEC), que es un desenlace compuesto que incluye diferentes variables (deterioro de capacidad de ejercicio, hospitalización por hipertensión arterial pulmonar, necesidad de aumentar el tratamiento para hipertensión arterial pulmonar o desenlaces combinados)2.
Los estudios a largo plazo fundamentados en eventos o desenlaces desarrollados recientemente incluyen COMPASS-2, AMBITION, SERAPHIN y GRIPHON. Los dos primeros evaluaron estrategias de tratamiento con medicamentos aprobados y los otros dos evaluaron nuevos medicamentos2.
Sin embargo, estos tratamientos disponibles solo mejoran parcialmente los síntomas y la sobrevida de los pacientes, razón por la cual se requiere un mejor entendimiento de la fisiopatología de la enfermedad, lo cual permitirá desarrollar nuevos tratamientos o intervenciones en estos pacientes1.
Fisiopatología: nuevos aspectosEl remodelamiento reverso de la vasculatura pulmonar es el origen del aumento de la presión pulmonar en la hipertensión arterial pulmonar, lo cual conlleva deterioro funcional progresivo, a pesar del tratamiento farmacológico disponible1,2.
Aunque las diferentes formas de hipertensión arterial pulmonar podrían reflejar distintos mecanismos fisiopatológicos, la evidencia actual sugiere que un denominador común para las alteraciones moleculares y celulares es el entrecruzamiento celular con la pared vascular. El entrecruzamiento entre las células endoteliales disfuncionales y otros componentes de la pared vascular pulmonar, como células musculares lisas, miofibroblastos y células inmunes circulantes, representa una característica importante de la patogénesis de la hipertensión arterial pulmonar1.
Otras lesiones vasculares pulmonares que se dan en pacientes con hipertensión arterial pulmonar incluyen la muscularización anormal de arterias precapilares mediales y distales, la pérdida de arterias precapilares, el engrosamiento de la pared arteriolar pulmonar con lesiones laminares excéntricas o concéntricas, la formación neointimal, la necrosis fibrinoide y finalmente la formación de lesiones vasculares complejas o “lesiones plexiformes”1.
Las bases moleculares y celulares de este remodelamiento vascular en pacientes con hipertensión arterial pulmonar se pueden clasificar según los mecanismos fisiopatológicos implicados1:
- 1.
Restauración del entrecruzamiento funcional celular entre células de la pared vascular: el endotelio pulmonar en la hipertensión arterial pulmonar es una fuente importante de mediadores claves para el remodelamiento vascular, tales como factores de crecimiento (FCF-2, serotonina, angiotensina), péptidos vasoactivos (ON, PGI-2, ET-1), citoquinas (IL-1, IL-6), quemoquinas (proteína quimioatrayente de monocitos) y adipoquinas (leptina). La sobreproducción endocrina de estos mediadores se asocia con aumento de la proliferación, sobrevida, migración y diferenciación celular vascular pulmonar.
- 2.
Corrección de procesos inflamatorios e inmunes inapropiados o alterados: los niveles circulantes de algunas citoquinas y quemoquinas se encuentran demasiado elevados y algunos se correlacionan con un peor curso clínico en pacientes con hipertensión arterial pulmonar. Desde el punto de vista histopatológico, las lesiones vasculares pulmonares en pacientes con hipertensión arterial pulmonar se caracterizan por diferentes grados de infiltrado inflamatorio perivascular compuesto por linfocitos T y B, macrófagos, células dendríticas y mastocitos.
- 3.
Restauración de un balance adecuado entre la síntesis y degradación de la matriz extracelular (MEC): los cambios cuantitativos y cualitativos en la MEC se asocian con un micromedio ambiente local aberrante en la pared vascular pulmonar remodelada en pacientes con hipertensión arterial pulmonar, creando entonces un entorno pericelular/extracelular que predispone a la proliferación, sobrevida y migración celular. El remodelamiento alterado de MEC puede promover el remodelamiento vascular pulmonar local de tres formas:
- a.
Generación de fragmentos de MEC que modulan directamente la proliferación, migración y activación de proteasas.
- b.
Liberación excesiva de factores de crecimiento y de varias moléculas incorporadas a la MEC.
- c.
Exposición de sitios funcionalmente importantes a nivel de colágeno, laminina, elastina o fibronectina.
- a.
- 4.
Restitución homeostática del sistema de señalización BMPRII/KCNK3: la mutación del gen BMPR2 es el principal factor de riesgo para hipertensión arterial pulmonar hereditaria. La mutación del gen KCNK3 también se ha relacionado con hipertensión arterial pulmonar hereditaria. Las mutaciones a lo largo del gen BMPR2 (a excepción del exón 13) se presentan como defectos de duplicación, deleción, duplicación y mutaciones missense y nonsense. De otro lado, todas las mutaciones identificadas en el gen KCNK3 corresponden a missense.
La hipertensión arterial pulmonar es una enfermedad compleja, con múltiples etiologías, y es mediada por una interacción de fenómenos genéticos, factores ambientales patológicos y cambios epigenéticos, lo cual explica la gran variabilidad en la susceptibilidad. Los fenómenos genéticos y los factores ambientales han sido ampliamente estudiados, mientras que los mecanismos epigenéticos, que hacen referencia a todos los cambios adquiridos en la expresión genética y que no se relacionan con cambios en la secuencia basal de ADN, son objeto de investigación en la actualidad. Los tres principales tipos de regulación epigenética son: 1) Metilación de ADN, 2) Modificación de histonas y 3) Micro ARN (MiARN)3.
Dentro de las posibles alteraciones epigenéticas de los genes relacionados con hipertensión arterial pulmonar se incluyen3:
- •
BMPR1
- •
BMPR2
- •
Endoglina: correceptor de señalización de BMP/ TGF-B
- •
SMADs: mediador principal de señalización de BMP
- •
Caveolina 1
- •
KNCK3: subfamilia de canal de potasio miembro K 3. tablas 1-3.
Tabla 1.Disfunción endotelial
Vasoconstricción sostenida Disminución de óxido nítrico Disminución de prostaglandina I2 Aumento de endotelina-1 Aumento de serotonina (5-HT) Aumento de angiotensina II Hiperproliferación Aumento de factores externos de crecimiento Factor de crecimiento de fibroblastos – 2 Factor de crecimiento derivado de plaquetas Factor de crecimiento epidérmico Disminución de proteínas proapoptóticas Célula mieloide -1 con secuencia de leucemia (Mcl-1) Células B linfoma – 2 (Bcl2) Célula mieloide -1 con secuencia de leucemia (Mcl-1) Hipercoagulación Aumento de factor Von Willebrand Aumento de trombomodulina Aumento de selectina P Aumento de tromboxano A2 Tabla 2.Hiperplasia de músculo liso
Hiperproliferación y resistencia a la apoptosis Aumento de factores de crecimiento exógenos Factor de crecimiento de fibroblastos – 2 Factor de crecimiento derivado de plaquetas Factor de crecimiento epidérmico Disminución de la vía BMPR2 Aumento de proteínas antiapoptóticas Aumento de migración Hiperpolarización Disminución de canales de potasio sensibles al ácido relacionado con TWIK TASK-1 Disminución de canales de potasio mediados por voltaje Aumento de receptor potencial transitorio 1 y 6 (TRPC1 y TRPC6) Aumento de receptores sensibilizantes de Ca (CaSr) Alteración de la energética celular Fragmentación mitocondrial e hiperpolarización en células musculares lisas Cambio glicolítico Tabla 3.Inflamación y alteración inmune
Inflamación sostenida Aumento de citoquinas y quemoquinas Il-1B, Il-6, MCP-1, CCL5, IL-13, CX3CL1 Infiltración celular inmune Macrófagos y monocitos Células citotóxicas – Natural killer Granulocitos Mastocitos Alteración inmune Células dendríticas, linfocitos T y B, alteraciones celulares de linfocitos T - Natural killer Autoinmunidad: balance alterado entre linfocitos T, presencia de autoanticuerpos Neogénesis linfoide
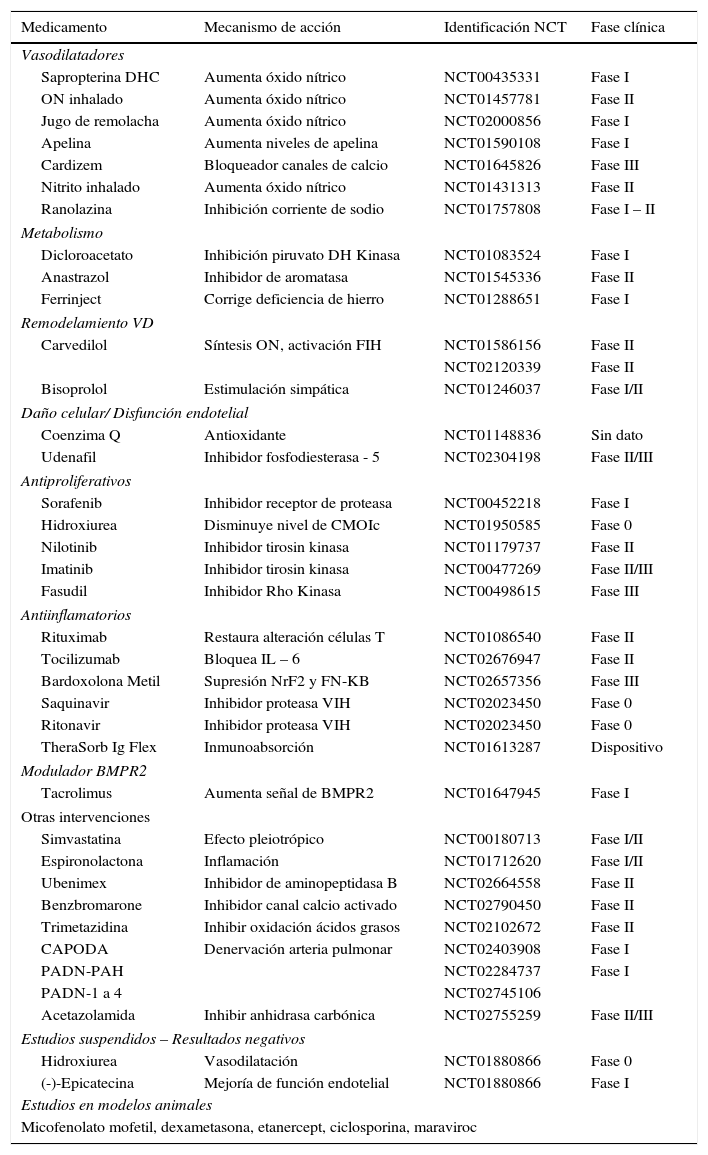
Nuevas opciones terapéuticas (tablas 4 y 5)
Hipertensión arterial pulmonar1,4–9
| Medicamento | Mecanismo de acción | Identificación NCT | Fase clínica |
|---|---|---|---|
| Vasodilatadores | |||
| Sapropterina DHC | Aumenta óxido nítrico | NCT00435331 | Fase I |
| ON inhalado | Aumenta óxido nítrico | NCT01457781 | Fase II |
| Jugo de remolacha | Aumenta óxido nítrico | NCT02000856 | Fase I |
| Apelina | Aumenta niveles de apelina | NCT01590108 | Fase I |
| Cardizem | Bloqueador canales de calcio | NCT01645826 | Fase III |
| Nitrito inhalado | Aumenta óxido nítrico | NCT01431313 | Fase II |
| Ranolazina | Inhibición corriente de sodio | NCT01757808 | Fase I – II |
| Metabolismo | |||
| Dicloroacetato | Inhibición piruvato DH Kinasa | NCT01083524 | Fase I |
| Anastrazol | Inhibidor de aromatasa | NCT01545336 | Fase II |
| Ferrinject | Corrige deficiencia de hierro | NCT01288651 | Fase I |
| Remodelamiento VD | |||
| Carvedilol | Síntesis ON, activación FIH | NCT01586156 | Fase II |
| NCT02120339 | Fase II | ||
| Bisoprolol | Estimulación simpática | NCT01246037 | Fase I/II |
| Daño celular/ Disfunción endotelial | |||
| Coenzima Q | Antioxidante | NCT01148836 | Sin dato |
| Udenafil | Inhibidor fosfodiesterasa - 5 | NCT02304198 | Fase II/III |
| Antiproliferativos | |||
| Sorafenib | Inhibidor receptor de proteasa | NCT00452218 | Fase I |
| Hidroxiurea | Disminuye nivel de CMOIc | NCT01950585 | Fase 0 |
| Nilotinib | Inhibidor tirosin kinasa | NCT01179737 | Fase II |
| Imatinib | Inhibidor tirosin kinasa | NCT00477269 | Fase II/III |
| Fasudil | Inhibidor Rho Kinasa | NCT00498615 | Fase III |
| Antiinflamatorios | |||
| Rituximab | Restaura alteración células T | NCT01086540 | Fase II |
| Tocilizumab | Bloquea IL – 6 | NCT02676947 | Fase II |
| Bardoxolona Metil | Supresión NrF2 y FN-KB | NCT02657356 | Fase III |
| Saquinavir | Inhibidor proteasa VIH | NCT02023450 | Fase 0 |
| Ritonavir | Inhibidor proteasa VIH | NCT02023450 | Fase 0 |
| TheraSorb Ig Flex | Inmunoabsorción | NCT01613287 | Dispositivo |
| Modulador BMPR2 | |||
| Tacrolimus | Aumenta señal de BMPR2 | NCT01647945 | Fase I |
| Otras intervenciones | |||
| Simvastatina | Efecto pleiotrópico | NCT00180713 | Fase I/II |
| Espironolactona | Inflamación | NCT01712620 | Fase I/II |
| Ubenimex | Inhibidor de aminopeptidasa B | NCT02664558 | Fase II |
| Benzbromarone | Inhibidor canal calcio activado | NCT02790450 | Fase II |
| Trimetazidina | Inhibir oxidación ácidos grasos | NCT02102672 | Fase II |
| CAPODA | Denervación arteria pulmonar | NCT02403908 | Fase I |
| PADN-PAH | NCT02284737 | Fase I | |
| PADN-1 a 4 | NCT02745106 | ||
| Acetazolamida | Inhibir anhidrasa carbónica | NCT02755259 | Fase II/III |
| Estudios suspendidos – Resultados negativos | |||
| Hidroxiurea | Vasodilatación | NCT01880866 | Fase 0 |
| (-)-Epicatecina | Mejoría de función endotelial | NCT01880866 | Fase I |
| Estudios en modelos animales | |||
| Micofenolato mofetil, dexametasona, etanercept, ciclosporina, maraviroc | |||
FIH: factor inducible de hipoxia. MOIc: células de médula ósea inmadura circulantes
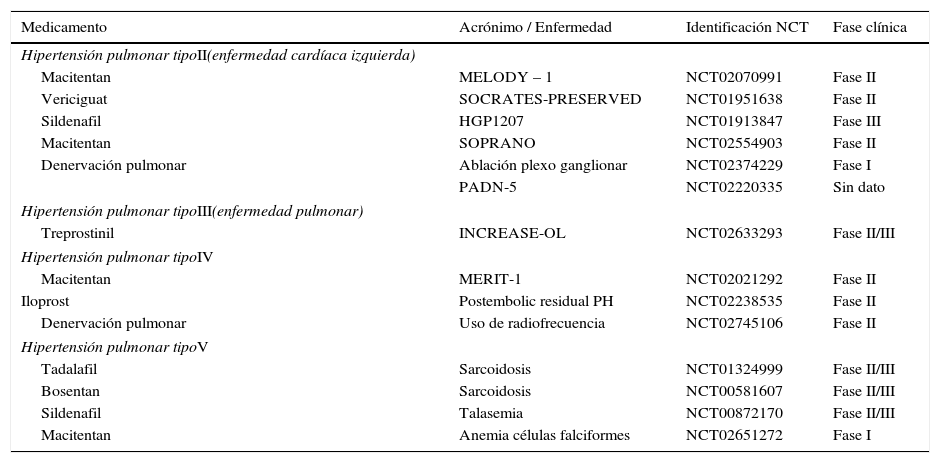
Hipertensión pulmonar tipos II, III, IV Y V2,8,9
| Medicamento | Acrónimo / Enfermedad | Identificación NCT | Fase clínica |
|---|---|---|---|
| Hipertensión pulmonar tipoII(enfermedad cardíaca izquierda) | |||
| Macitentan | MELODY – 1 | NCT02070991 | Fase II |
| Vericiguat | SOCRATES-PRESERVED | NCT01951638 | Fase II |
| Sildenafil | HGP1207 | NCT01913847 | Fase III |
| Macitentan | SOPRANO | NCT02554903 | Fase II |
| Denervación pulmonar | Ablación plexo ganglionar | NCT02374229 | Fase I |
| PADN-5 | NCT02220335 | Sin dato | |
| Hipertensión pulmonar tipoIII(enfermedad pulmonar) | |||
| Treprostinil | INCREASE-OL | NCT02633293 | Fase II/III |
| Hipertensión pulmonar tipoIV | |||
| Macitentan | MERIT-1 | NCT02021292 | Fase II |
| Iloprost | Postembolic residual PH | NCT02238535 | Fase II |
| Denervación pulmonar | Uso de radiofrecuencia | NCT02745106 | Fase II |
| Hipertensión pulmonar tipoV | |||
| Tadalafil | Sarcoidosis | NCT01324999 | Fase II/III |
| Bosentan | Sarcoidosis | NCT00581607 | Fase II/III |
| Sildenafil | Talasemia | NCT00872170 | Fase II/III |
| Macitentan | Anemia células falciformes | NCT02651272 | Fase I |
- •
Modulación de HDAC (histone deacetylase)
- •
Inhibición de amplio espectro de HDAC: tricostatina A
- •
Inhibición selectiva de HDAC clase II
- •
Inhibidor de DNMT (DNA methyltransferase): azacitidina – 5
- •
Inhibidor de MiARN: miravirsen, MRX34
Desde el descubrimiento de la asociación entre la hipertensión arterial pulmonar hereditaria y las mutaciones en el gen-2 del receptor de la proteína morfogénica del hueso (BMPR2) se ha avanzado mucho en la comprensión de las bases genéticas de la hipertensión arterial pulmonar. Recientemente se han descrito nuevas asociaciones entre ésta y mutaciones en otros genes, como caveolina – 1 y KCNK3. Estos avances han permitido el desarrollo de la tecnología genómica para personalizar el tratamiento especializado y mejorar las herramientas para predecir el desarrollo de la enfermedad en portadores susceptibles4.
El paciente con hipertensión arterial pulmonar deben ser evaluado con el objetivo de descartar formas hereditarias o familiares y considerar estudios genéticos en aquellos con un alto índice de sospecha. Así mismo, es pertinente tener en cuenta las implicaciones éticas y legales de la identificación de mutaciones genéticas específicas y del riesgo de desarrollar hipertensión arterial pulmonar antes de realizar estas pruebas4.
Conflicto de interesesNinguno.