


INTRODUCCIÓN
El síndrome XYY es una alteración genética, y es una de las anomalías cromosómicas más frecuentes en la población masculina. Fue descrito por primera vez en la década de los sesenta por P. Jacobs, quien estudió a sujetos internados por su propensión a la violencia y criminalidad; su incidencia es de entre 1/1.000 y 1/700 varones. Se caracteriza por una trisomía, se encuentra un cromosoma Y extra en el cariotipo de estos varones. Puede presentarse de forma completa, es decir, en todas las células, o en forma de mosaico (46, XY/47, XYY). Por otro lado, se han descrito 2 mecanismos principales que originarían esta alteración: a) sin disyunción paterna en meiosis II durante gametogénesis, y b) error mitótico poscigótico (originándose mosaicismo según la etapa de no disyunción mitótica), el primero es el más frecuente1.
Su diagnóstico suele ser tardío, ya que por lo general no presentan alteraciones fenotípicas, y una causa frecuente de estudio diagnóstico es la consulta por infertilidad2. Los rasgos que hay que destacar son talla alta, mayor fuerza, trastornos de aprendizaje y lenguaje, y tendencia a la agresividad. Además se ha asociado a una mayor tasa de criminalidad, y se han descrito incidencias más altas de este síndrome en recintos carcelarios. Sin embargo, últimamente esto ha sido tema de controversia3.
En cuanto a su fertilidad, gran parte de los casos es fértil, con normozoospermia en el análisis del seminograma. Se ha postulado que en estos casos se perdería el cromosoma Y extra previo a la meiosis en la formación de gametos. Sin embargo, hay casos de pacientes con oligoastenozoospermia severa y azoospermia. En ellos persistiría el cromosoma Y extra en profase meiótica, con aumento de la degeneración espermática y, por tanto, causando infertilidad4.
CASO CLÍNICO
Se presenta el caso de una pareja que consulta por esterilidad primaria de 4 años de evolución. Esposa de 34 años sin factores patológicos de infertilidad. Varón de 35 años de edad sin estudios de fertilidad previos. Como antecedentes personales de interés, en el varón hay que destacar: dificultad a la expresión oral e inadaptación social y familiar, que requiere tratamiento con ansiolíticos, con buena respuesta (alprazolam 0,5 mg/12 h y diazepam 10 mg/12 h). Sin otras patologías de interés.
Entre los antecedentes familiares hay que destacar que el paciente refiere "tío y primo muy nerviosos que requieren tratamiento psiquiátrico", sin tener más conocimiento o detalles mayores de la patología exacta que tienen y sin haber sido estudiados desde el punto de vista genético.
A la exploración física del paciente se encontró: talla 180 cm; peso 78 kg; índice de masa corporal 27; sin otros aspectos destacables en el examen físico general ni segmentario.
Se le realizaron las siguientes pruebas complementarias:
Dos seminogramas, en los que se comprobó una oligoastenoteratozoospermia (OAT) severa. Volumen: 2 ml. Recuento: 4,5 millones de espermatozoides por ml. Total: 14 millones. Motilidad: 0% GIII, 15% GII, 50% GI y 35% inmóviles. Morfología: 2% formas normales. Vitalidad: 15% formas vivas.
Analítica para determinación hormonal, que estaba dentro de rangos normales: FSH, LH, testosterona total y prolactina.
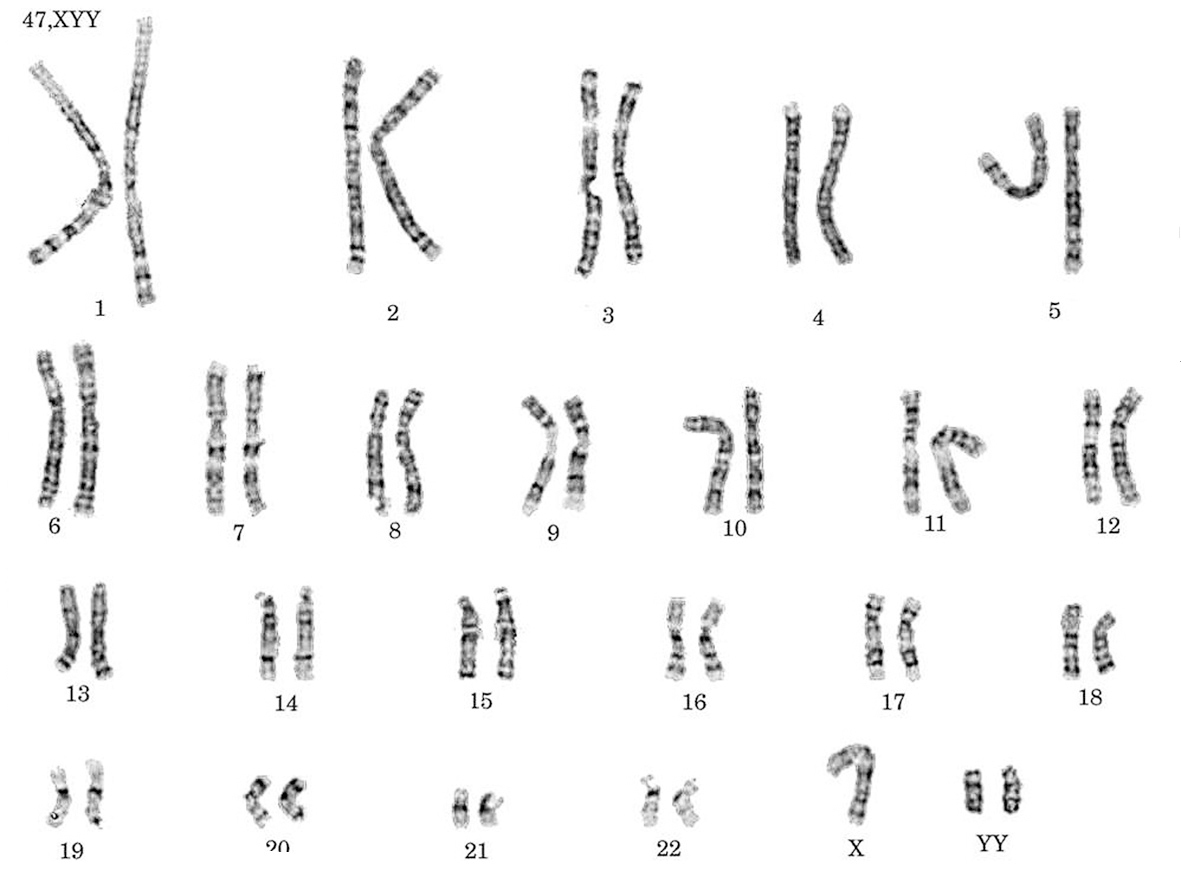
Cariotipo: 47, XYY (fig. 1).
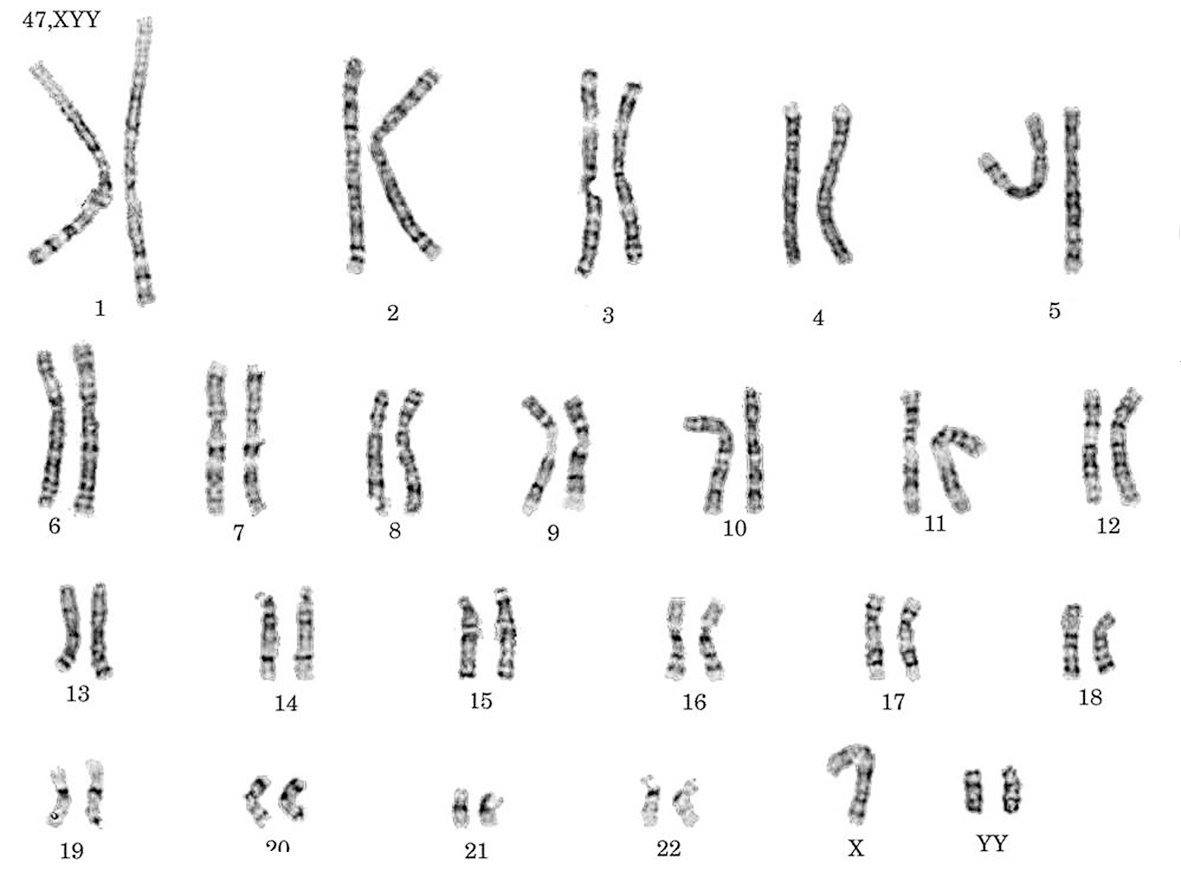
Figura 1. Cariotipo de un varón con síndrome 47, XYY.
Biopsia testicular para estudio de meiosis: destacan anomalías de apareamiento con desinapsis que pueden producir espermatozoides aneuploides.
Ante los resultados complementarios obtenidos se lleva a cabo consejo genético y se ofrecen las diferentes opciones existentes: a) no realizar nada más; b) adopción, y c) técnicas de reproducción asistida, realizando diagnóstico genético preimplantacional previo a la transferencia de embriones, seleccionando así los que presenten un cariotipo normal.
En este caso la pareja opta por la adopción con resultado satisfactorio.
DISCUSIÓN
Como ya se ha mencionado, en el síndrome 47, XYY, se pueden presentar: a) varones en los que se ha perdido el cromosoma Y extra en meiosis durante la gametogénesis, por tanto presentan normozoospermia, y son fértiles, y b) varones en los que persisten gametos con este cromosoma, con OAT severa o azoospermia, en cuyo caso serían infértiles1,5.
Habiendo descrito ya el mecanismo por el cual se produce esta alteración, se ahondará en el enfoque terapéutico de pacientes con este síndrome y que consultan por infertilidad. En los varones con cariotipo 47, XYY en los que se produce una ausencia de disyunción meiótica en paquitene durante la gametogénesis, se origina una OAT severa, con la consecuente infertilidad. En el contexto de estas parejas que consultan por infertilidad, es fundamental realizar un consejo genético y mostrar las diferentes alternativas por las que pueden optar, entendiendo siempre que lograr ser padres no es un deber, por lo que no se debe forzar a las parejas a incurrir en técnicas que no deseen.
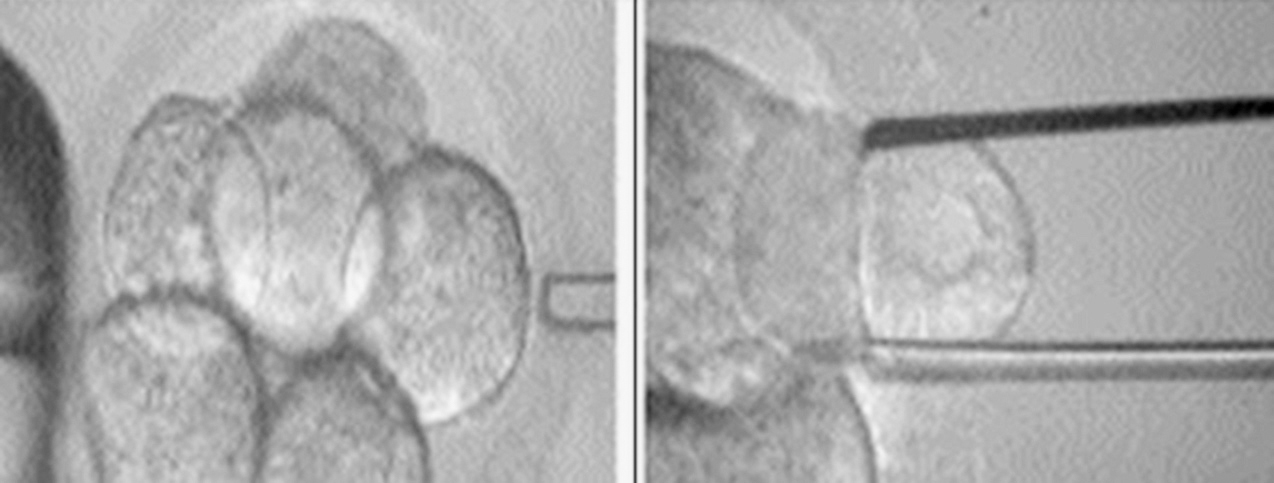
Ante la producción de gametos con un cromosoma Y extra, se corre el riesgo de transmitir a la descendencia dicha alteración genética, por lo que resulta fundamental explicar este hecho a los padres antes de buscar el embarazo. Actualmente, con los avances tecnológicos y el desarrollo de las técnicas de reproducción asistida se abre la posibilidad a las parejas que lo deseen de realizar diagnóstico genético preimplantacional (DGP) del embrión ante casos de alteraciones genéticas, como lo es el caso en cuestión. Esta técnica busca el análisis genético de embriones producto de técnicas de reproducción asistida, con la posterior transferencia de los caracterizados como sanos, así se evita la persistencia de ciertas anomalías genéticas transmisibles por los padres6. Iniciado en la década de los noventa, este procedimiento ha revolucionado el desarrollo de las técnicas de reproducción asistida, en cuanto permite tener hijos cromosómicamente normales a los padres con riesgo de transmitir anomalías genéticas a su descendencia. Sin embargo, cabe destacar que, aunque ciertas parejas pudiesen lograr un embarazo natural, para realizar el DGP es absolutamente necesario obtener embriones a través de técnicas de fertilización in vitro, por lo que dentro del consejo genético debe explicarse a la pareja estas técnicas y las tasas de éxito que actualmente alcanzan, aproximadamente el 30-35%6, y que el éxito de la técnica también dependerá del número de embriones normales obtenidos y finalmente transferidos. Para el DGP se debe realizar biopsia embrionaria previa transferencia de éstos (fig. 2), la cual se hará en el día +3 posfecundación, momento en el que se obtienen suficientes células para estudio. En ciertos trabajos se planteó la posibilidad de que esta biopsia alterara la calidad del embrión, disminuyendo la tasa de éxito; sin embargo, con las mejorías en las técnicas de biopsia y ante la evidencia de que la tasa de gestaciones viables con DGP, en centros especializados, es cercana a la obtenida con técnicas de fertilización in vitro sin este diagnóstico, se puede proponer esta técnica sin el riesgo de que empeore la tasa de éxito de la técnica de reproducción asistida utilizada7.
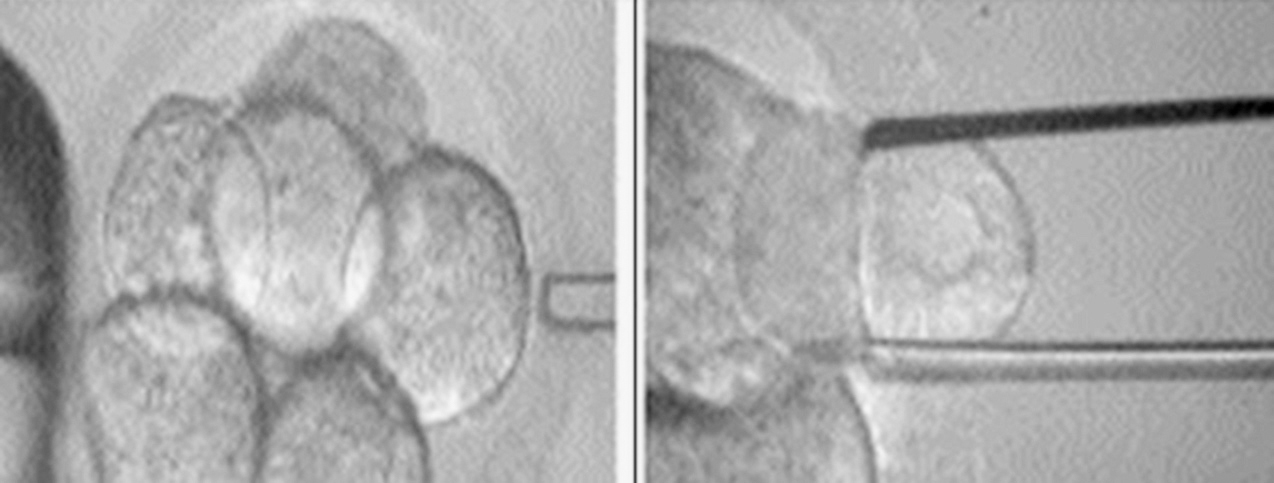
Figura 2. Imagen que muestra el procedimiento de biopsia de células embrionarias para realizar estudio citogenético previo a su transferencia (Muñoz-Núñez7).
Las técnicas más utilizadas para realizar el DGP son la PCR, la de hibridación in situ fluorescente (FISH), la electroforesis, la citometría de flujo y el análisis de cariotipo; actualmente, la más utilizada en análisis de anomalías cromosómicas relacionadas con los genes X e Y es la FISH7.
Sin embargo, aunque con baja frecuencia, el DGP presenta errores diagnósticos, cuyo porcentaje varía según la técnica utilizada, para la FISH es de aproximadamente un 5%8. Por lo tanto, a las parejas que incurran en esta técnica se les debe recomendar de todas formas el DGP mediante amniocentesis para confirmar la normalidad genética del embrión en curso.
CONCLUSIÓN
Habiendo presentado el caso clínico, y tras la revisión bibliográfica expuesta, se puede concluir que ante parejas que consultan por infertilidad sin factor femenino presente es fundamental el estudio de cariotipo del varón, aunque no se tengan antecedentes de alteraciones genéticas, dada la baja frecuencia de diagnóstico temprano de este síndrome. Por otra parte, confirmada esta alteración es fundamental realizar el respectivo consejo genético a la pareja, brindando la información necesaria para la adecuada decisión de ésta. Dado el avance tecnológico en las técnicas de reproducción asistida, se propone la fertilización in vitro con DGP y posterior amniocentesis en parejas que busquen fertilidad y en las cuales el varón presente el cariotipo 47, XYY. En caso de que la pareja no desee practicar técnicas de reproducción asistida se debe proponer, siendo igualmente válido, optar por no realizar mayores tratamientos o ir a adopción, como fue la resolución del caso clínico expuesto.
Correspondencia: Dra. M.J. del Río.
IANDROMS. Paseo de la Bonanova, 69, 1.ª planta.
08017 Barcelona. España.
Correo electrónico: iandroms@iandroms.com