





El desarrollo normal del esqueleto requiere de la existencia de movimientos fetales normales en frecuencia e intensidad. Cualquier restricción al desplazamiento normal en el feto en desarrollo, ya sea por factores intrínsecos o extrínsecos como los producidos por trastornos de las motoneuronas, músculos, sistema nervioso central, tejido conectivo, ambiente uterino, toxinas exógenas, afectará al feto. En esta revisión se resumen las manifestaciones clínicas, el abordaje diagnóstico y las diversas etiologías subyacentes a las contracturas articulares múltiples en el feto, haciendo hincapié en el espectro cada vez mayor de enfermedades genéticas específicamente en el campo neuromuscular. Los avances experimentados en las imágenes prenatales y las nuevas herramientas de genética molecular han permitido alcanzar un diagnóstico etiológico en un número cada vez mayor de pacientes, otorgar un mejor asesoramiento genético a la familia así como preparar las condiciones de tratamiento más favorables para el recién nacido.
Normal fetal movements are essential to skeletal development. Any restriction to normal movement on the developing fetus, either by intrinsic or extrinsic factors, ie disorders of motor neuron, muscle, central nervous system, connective tissue, uterine ambiance, exogenous toxics, will affect the fetus. At the most severe end, the fetal akinesia deformation sequence (FADS) occurs with highly abnormal or absent fetal movement while a substantial reduction in fetal movement can lead to multiple arthrogryposis. In this review we summarized the diverse etiologies underlying FADS and arthrogryposis, emphasizing the ever-expanding spectrum of genetic conditions specifically on neuromuscular field. Even if there is still almost half of time, uncertainty about the final etiology, the progress on prenatal fetal imaging and molecular genetic tools, will allow to give a better genetic counseling to the family and prepare the most favorable treatment conditions for the upcoming newborn.
La akinesia fetal es un término descriptivo para referirse a un espectro amplio de trastornos que tienen en común el desarrollo de contracturas asociadas a la disminución o ausencia de movimientos fetales. En la literatura médica se han utilizado diferentes nombres para referirse a esta condición, tales como secuencia de deformación de akinesia/hipokinesia fetal (FADS), secuencia de Hipokinesia Fetal (HF), síndrome de Pena-Shokeir, amioplasia congénita, Artrogriposis Múltiple Congénita (AMC), síndrome contractural letal congénito y el síndrome de pterigium múltiple (MPS). El factor común en todas ellas, es la hipomotilidad del feto, secundaria a un numeroso espectro de etiologías genéticas y ambientales 1.
Pese a que la akinesia fetal es una entidad rara, se han comunicado incidencias de artrogriposis de hasta 1:3000 a 1:5000 recién nacidos vivos, siendo la más frecuente la amioplasia congénita, con una frecuencia de 1:10000 RNV 2. Es interesante conocer así mismo, que el 1% de los niños tiene al nacer algún tipo de contractura, incluido el pie bot, que tiene una incidencia de 1/300 nacimientos y la camptodactilia que se presenta en 1/200 3. En Chile la tasa de incidencia de artrogriposis es de 1,6 por 10000 nacidos vivos y la de pie bot es de 1/625, ambas cifras muy semejantes al registro del Estudio Latino Americano de Malformaciones Congénitas (ECLAM) del cual Chile forma parte desde el año 1969 4.
En términos generales las contracturas en la akinesia son producto de una disminución de los movimientos fetales, secundarios a su vez a alteraciones en el sistema nervioso central, nervios periféricos, músculos, tejido conectivo, enfermedades maternas, agentes ambientales y/o compromiso vascular. Esta entidad es por lo tanto una ventana que pone en evidencia todos aquellos factores en el desarrollo embrionario, imprescindibles para lograr un adecuado movimiento in útero que permitirá el desarrollo normal de las articulaciones del feto.
En los últimos 30 años ha habido un gran progreso en distinguir distintos tipos específicos de akinesia fetal, con más de 400 enfermedades identificadas que desarrollan artrogriposis como manifestación clínica y más de 150 mutaciones genéticas 1. Situación que añade complejidad al desafío de establecer la etiología subyacente en cada caso particular. Ha sido gracias al desarrollo de la genética molecular, con la implementación de las técnicas de hibridación genómica comparada (CGH array), secuenciación masiva de nueva generación (next generation sequencing) exoma, (whole exome sequencing), genoma (whole genome scan) y otros, lo que ha permitido el extraordinario progreso en la identificación de causas específicas de las distintas artrogriposis5,6. Sin embargo, en un porcentaje significativo de pacientes, aún no es posible establecer un diagnóstico genético específico 5.
La efectividad del diagnóstico prenatal en la HF sigue siendo un desafío, ya que estudios retrospectivos de registros ultrasonográficos de pacientes con amioplasia, han demostrado una falla en la detección prenatal de esta condición de hasta un 75% de los casos 7. Es por lo tanto un desafío para el clínico y los profesionales de la salud la pesquisa prenatal y el establecimiento de la etiología subyacente a los cuadros de artrogriposis de manera de ofrecer las mejores opciones de nacimiento para el feto en gestación, preparar a la familia y optimizar el manejo postnatal del recién nacido.
Este artículo tiene como propósito otorgar una aproximación general a la akinesia/hipokinesia fetal, a su clasificación, etiología, forma de aproximación clínica y algoritmo de estudio, con énfasis en etiologías neuromusculares e indicadores de sospecha diagnóstica precoz a través de las diferentes técnicas de imágenes.
METODOLOGIAEn este artículo de revisión se realizó una búsqueda de artículos en las bases Pubmed, SciELO, que contuvieran las siguientes palabras claves: fetal akinesia, fetal hipokinesia, arthrogryposis, fetal akinesia deformation sequencing, Pena Shokeir, fetal hypomobility syndrome, neuromuscular, congenital myopathy, congenital motor neuron, congenital myasthenia, congenital contractural syndrome, amyoplasia, prenatal diagnosis. Se restringió la búsqueda a los últimos 30 años, desde el 1985 hasta el 2015. Se consideraron principalmente aquellos artículos originales, epidemiológicos y de revisión.
DEFINICIONESSecuencia de deformación akinésica/hipokinesia fetalLa secuencia de deformación akinésica del feto (FADS) es un término descriptivo que integra las consecuencias en el feto de la limitación al movimiento intrauterino y por ende comprende numerosas y heterogéneas causas responsables de ella. Su incidencia se estima en 1:150000 8. El movimiento fetal comienza a las 8 semanas de gestación. El desarrollo normal de las articulaciones requiere una movilización de éstas con un rango completo de movimiento. Mientras más precoz sea la falta de movilización adecuada del feto, más grave es la artrogriposis. Las contracturas articulares se producen por el engrosamiento de la capsula articular y tejido circundante, secundaria a una respuesta anormal del colágeno a la falta de movimiento. Adicionalmente, la ausencia de movimientos torácicos rítmicos, que simulan una respiración en la edad fetal, determinan un crecimiento inadecuado de la caja torácica, inmadurez de los alvéolos y surfactante y una hipoplasia pulmonar9,10.
Las siguientes son las manifestaciones clínicas encontradas en FADS:
- •
Contracturas articulares múltiples
- •
Pterigium en las extremidades
- •
Hipoplasia pulmonar
- •
Cordón umbilical corto
- •
Retardo del crecimiento intrauterino
- •
Intestino corto
- •
Líquido amniótico anormal: polihidroamnios por ausencia de deglución fetal
- •
Al nacer: fracturas por osteoporosis, anomalías craneofaciales como paladar hendido, cuello corto, orejas de implantación baja,
- •
Criptorquidea
- •
Inmadurez intestinal por dificultades en la alimentación
Este síndrome ha sido revisado recientemente de manera extensa y es la versión más severa de lo que hemos definido mas arriba como FADS 9. Es una entidad rara y letal, con una incidencia de 1:12000 y menos de 100 casos comunicados en la literatura. Se caracteriza por presentar camptodactilia e hipoplasia pulmonar severa, que ocasiona en la mayoría de estos individuos una muerte in útero. Existen varios subtipos que pueden o no acompañarse de malformaciones del sistema nervioso central como hidrocefalia, hipoplasia olivopontocerebelosa, hidranencefalia y otros. Las características craneofaciales relacionadas con la disminución del movimiento en este cuadro incluyen: hipertelorismo ocular, puente nasal alto, punta de la nariz escasamente desarrollada, orejas posteriores anguladas de implantación baja, cuello corto con pterigium leve, microretrognatia, boca pequeña, apertura mandibular limitada, paladar ojival, y con frecuencia paladar hendido submucoso 9.
Artrogriposis múltiple congénitaEl nombre artrogriposis deriva de la lengua griega arthron: articulación y gryposis: curvatura. Este nombre describe varias formas de contracturas congénitas de las extremidades o limitación en los rangos de movimientos pasivos y activos de las articulaciones acompañado de anomalías estructurales y/o funcionales de los tejidos blandos circundantes como la cápsula articular y ligamentos periarticulares. Dependiendo del momento en el desarrollo fetal en el que aparezca la hipomotilidad y la intensidad de ésta, será el grado de compromiso general del desarrollo esquelético y general del feto, variando desde una secuencia de deformación akinésica, cuando el movimiento fetal se ve entorpecido precozmente hasta una artrogriposis distal en aquellos casos más tardíos. Usamos el término artrogriposis para contracturas que afectan al menos a dos articulaciones en dos regiones diferentes del cuerpo. Estas contracturas son por lo general no progresivas y con frecuencia mejoran gradualmente con un apropiado y oportuno manejo con terapia física y ortopédica, pero que presentan tendencia a reproducirse y volver a la posición inicial si no se mantiene la terapia. El primer nombre que se le dio a esta condición en la literatura fue el de miodistrofia congénita en 1841, posteriormente se le llamó “contracturas congénitas múltiples” en 1897. El nombre “artrogriposis múltiple congénita” utilizado hasta la fecha fue acuñado por Stern en 192311.
AmioplasiaLa amioplasia es un subtipo específico de artrogriposis y la de mayor frecuencia, representando un 25-30% de todos los casos de artrogriposis múltiple congénita.
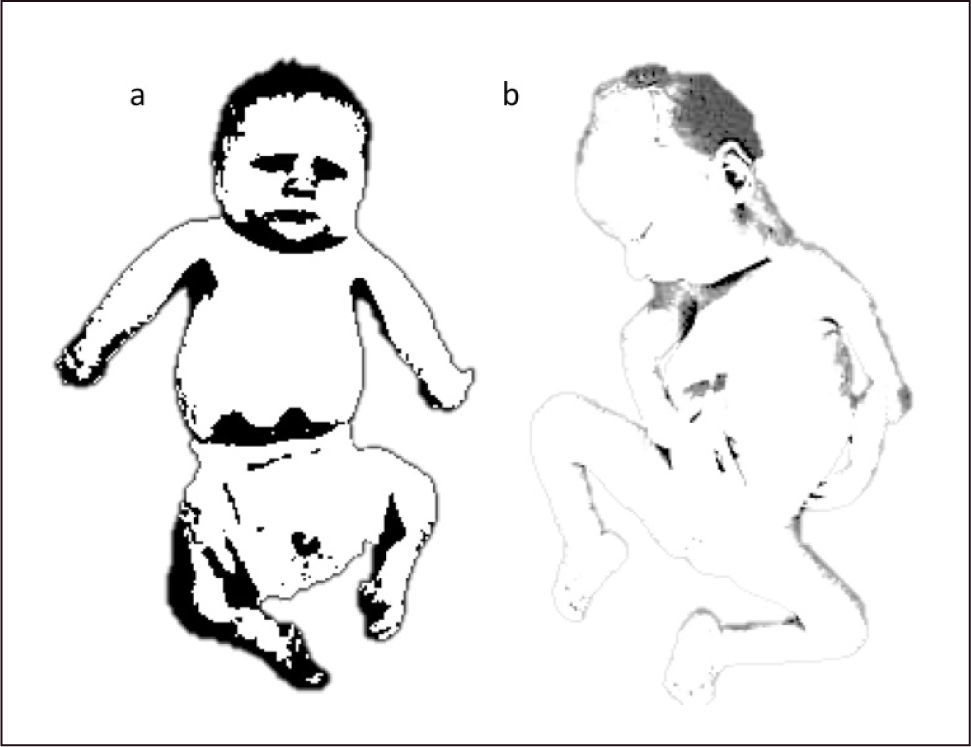
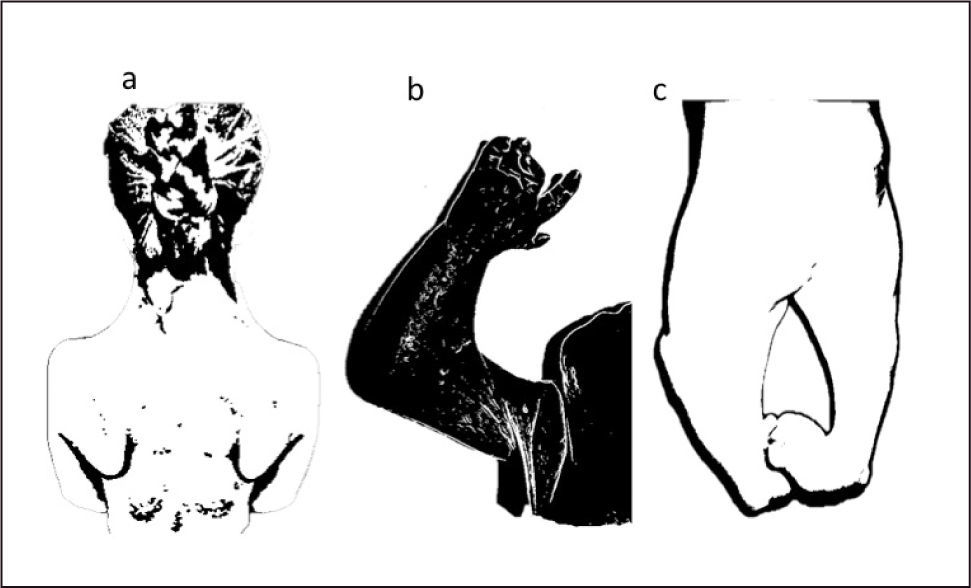
La amioplasia congénita es una condición esporádica, de etiología indeterminada, caracterizada por la presencia de un posicionamiento específico de las extremidades cuyos músculos han sido reemplazados por tejido fibroso y graso, afectando extremidades superiores, inferiores o ambas. Al nacer las extremidades superiores presentan una rotación interna en los hombros, los codos rígidos en extensión y flexión de las muñecas (Figura 1). En las extremidades inferiores es frecuente encontrar un equino varo grave de los pies, con contracturas en las rodillas y las caderas en variadas posiciones. La masa muscular está disminuida, existe un retraso del crecimiento intrauterino, acortamiento discreto de las extremidades y hoyuelos en las articulaciones afectadas. La inteligencia es normal y en general no hay otras malformaciones presentes salvo anomalías menores como dígitos hipoplásicos, hipoplasia del escroto o de los labios mayores12,13.

RECIÉN NACIDO CON AMIOPLASIA
a. Recién nacido con amioplasia. Compromiso simétrico de extremidades superiores e inferiores. Extremidades superiores con codos extendidos y rotación interna de hombros. Muñecas en flexión. Pies en equino-varo. b. Artrogriposis múltiple congénita. Deformación severa de articulaciones en extremidades superiores con flexión de codos y pie equino.
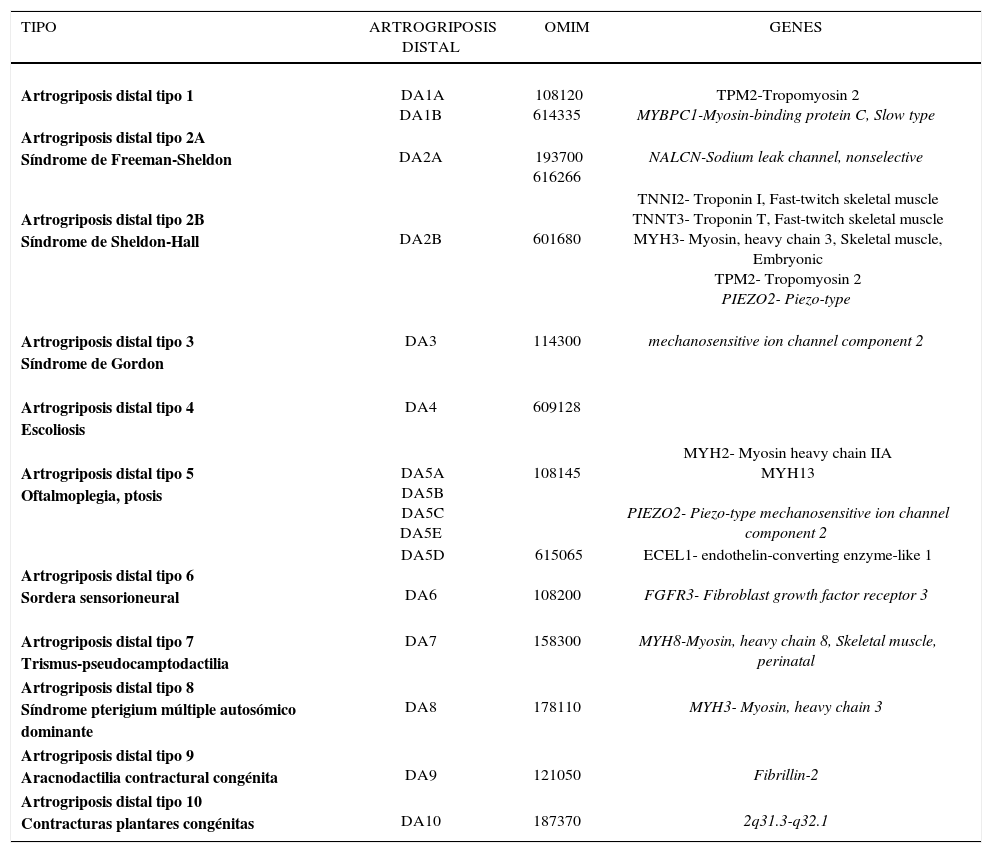
Las artrogriposis distales (AD) son un grupo de síndromes con contracturas que afectan primariamente manos y pies, sin afectar articulaciones proximales y frecuentemente asociadas a dismorfias faciales y herencia dominante. La incidencia en distintas series puede variar desde un 7 a un 35% del total de pacientes con artrogriposis sin embargo la prevalencia es desconocida. Las formas más comunes son la AD1 y la AD2B. La AD1 se caracteriza por la presencia de puños cerrados al nacer con desviación cubital, dedos superpuestos en sentido medial y pie equino u otras malas posiciones de los pies. Las caderas pueden estar afectadas, las pantorrillas delgadas y la apertura bucal limitada. La AD2A o síndrome de Freeman-Sheldon, se caracteriza por una boca pequeña, contracturas faciales y articulares distales, escoliosis y baja estatura La AD2B también llamado síndrome de Sheldon-Hall, es similar, pero menos severo que la tipo AD2A.
Los distintos tipos de artrogriposis distales se originan por mutaciones en diversas proteínas del sarcómero, que es la unidad funcional contráctil del músculo. Algunas mutaciones en la β tropomiosina TPM2, pueden ser responsable de la AD1, así como mutaciones en la troponina I, TNNI2 y troponina T, TNNT3 en la AD2B. Mutaciones en la cabeza pesada de la miosina embriónica pueden originar una AD2A y también la AD2B14–16. En la tabla 1 se detallan los diferentes tipos de artrogriposis distales, segun gen afectado.
ARTROGRIPOSIS DISTALES
| TIPO | ARTROGRIPOSIS DISTAL | OMIM | GENES |
|---|---|---|---|
Artrogriposis distal tipo 1 | DA1A DA1B | 108120 614335 | TPM2-Tropomyosin 2 MYBPC1-Myosin-binding protein C, Slow type |
| Artrogriposis distal tipo 2A Síndrome de Freeman-Sheldon | DA2A | 193700 616266 | NALCN-Sodium leak channel, nonselective |
Artrogriposis distal tipo 2B Síndrome de Sheldon-Hall | DA2B | 601680 | TNNI2- Troponin I, Fast-twitch skeletal muscle TNNT3- Troponin T, Fast-twitch skeletal muscle MYH3- Myosin, heavy chain 3, Skeletal muscle, Embryonic TPM2- Tropomyosin 2 PIEZO2- Piezo-type |
Artrogriposis distal tipo 3 Síndrome de Gordon | DA3 | 114300 | mechanosensitive ion channel component 2 |
Artrogriposis distal tipo 4 Escoliosis | DA4 | 609128 | |
Artrogriposis distal tipo 5 Oftalmoplegia, ptosis | DA5A DA5B DA5C DA5E | 108145 | MYH2- Myosin heavy chain IIA MYH13 PIEZO2- Piezo-type mechanosensitive ion channel component 2 |
Artrogriposis distal tipo 6 Sordera sensorioneural | DA5D DA6 | 615065 108200 | ECEL1- endothelin-converting enzyme-like 1 FGFR3- Fibroblast growth factor receptor 3 |
Artrogriposis distal tipo 7 Trismus-pseudocamptodactilia | DA7 | 158300 | MYH8-Myosin, heavy chain 8, Skeletal muscle, perinatal |
| Artrogriposis distal tipo 8 Síndrome pterigium múltiple autosómico dominante | DA8 | 178110 | MYH3- Myosin, heavy chain 3 |
| Artrogriposis distal tipo 9 Aracnodactilia contractural congénita | DA9 | 121050 | Fibrillin-2 |
| Artrogriposis distal tipo 10 Contracturas plantares congénitas | DA10 | 187370 | 2q31.3-q32.1 |
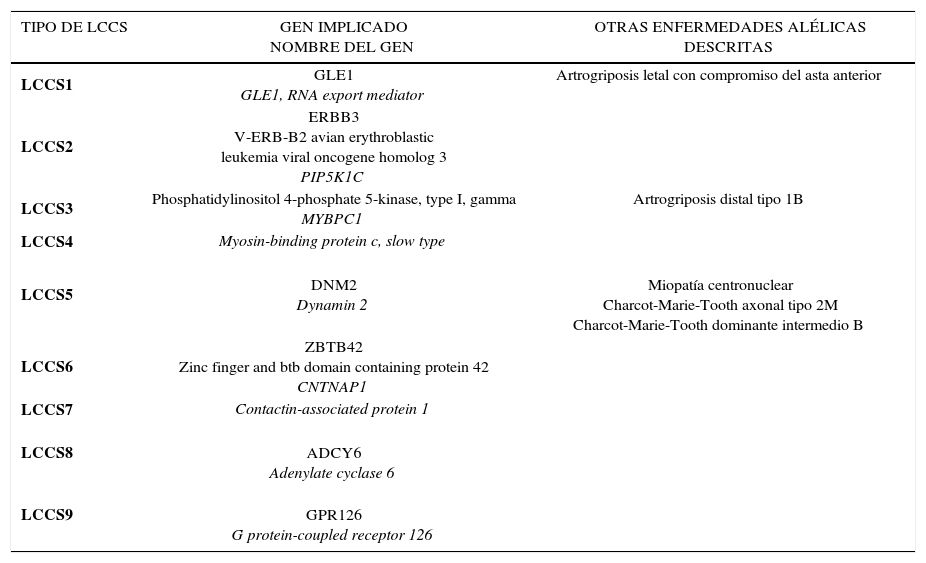
El síndrome contractural congénito letal (LCCS), entidad autosómica recesiva, es la forma más grave y siempre letal de la artrogriposis congénita múltiple. Es un trastorno caracterizado por contracturas articulares congénitas no progresivas que se asemeja en muchos aspectos al síndrome de Pena-Shokeir I, pero que difiere de éste por la no supervivencia después del nacimiento, por la presencia de hidrops fetal marcado y adelgazamiento generalizado de los huesos tubulares. Las contracturas pueden afectar las extremidades superiores o inferiores y/o la columna vertebral, dando lugar a diferentes grados de limitación de la flexión o extensión. El LCCS conduce a la muerte prenatal del feto antes de la trigésima segunda semana de gestación. El sello distintivo, altamente específico de LCCS es la degeneración de las neuronas motoras del asta anterior de la médula espinal 17.
Este síndrome presenta una alta heterogeneidad genética, distinguiéndose 9 tipos secundarios a mutaciones en distintos genes. (Tabla 2).
SÍNDROME CONTRACTURAL CONGÉNITO LETAL (LCCS)
| TIPO DE LCCS | GEN IMPLICADO NOMBRE DEL GEN | OTRAS ENFERMEDADES ALÉLICAS DESCRITAS |
|---|---|---|
| LCCS1 | GLE1 GLE1, RNA export mediator | Artrogriposis letal con compromiso del asta anterior |
| LCCS2 | ERBB3 V-ERB-B2 avian erythroblastic leukemia viral oncogene homolog 3 PIP5K1C | |
| LCCS3 | Phosphatidylinositol 4-phosphate 5-kinase, type I, gamma MYBPC1 | Artrogriposis distal tipo 1B |
| LCCS4 | Myosin-binding protein c, slow type | |
| LCCS5 | DNM2 Dynamin 2 | Miopatía centronuclear Charcot-Marie-Tooth axonal tipo 2M Charcot-Marie-Tooth dominante intermedio B |
| LCCS6 | ZBTB42 Zinc finger and btb domain containing protein 42 CNTNAP1 | |
| LCCS7 | Contactin-associated protein 1 | |
| LCCS8 | ADCY6 Adenylate cyclase 6 | |
| LCCS9 | GPR126 G protein-coupled receptor 126 |
Este trastorno fenotípica y genotípicamente heterogéneos se caracteriza por retraso del crecimiento, pliegues cutáneos anormales o pterigium múltiples, que comprometen el cuello, áreas antecubital, poplítea, intercrural, y los dedos, con contracturas en flexión de numerosas articulaciones (Figura 2). Además de anomalías genitales, paladar hendido y algunas ocasiones hipoplasia del corazón, pulmón, riñón o cerebro. Hay dos formas descritas de síndrome de pterigium múltiple (MPS), el síndrome de pterigium múltiple tipo Escobar, la forma más leve, también conocido como síndrome de Escobar (SE), y el síndrome pterigium múltiple letal. El síndrome de Escobar es secundario a una mutación homocigota o heterocigota compuesta en el gen CHRNG, que codifica la subunidad gamma del receptor de acetilcolina en el cromosoma 2q. La forma severa, que es fatal antes o poco después del nacimiento, tiene muchos de los mismos signos y síntomas que el SE. Se caracteriza por crecimiento prenatal insuficiente, pliegues dérmicos o pterigion presentes en múltiples áreas articulares y falta de movimiento de los músculos que conduce a una debilidad muscular y artrogriposis severa. Es causada por mutaciones en tres genes distintos, todos relacionados con la placa neuromuscular. Uno de ellos es el mismo gen implicado en el SE, el gen CHRNG. Así también se han descrito mutaciones en los genes CHRNA1 y CHRND que codifican para la subunidad alfa y delta del receptor de acetilcolina. Algunos de los síndromes miasténico congénitos de canal rápido o lento son causados por mutaciones en estos mismos genes18–21.
FISIOPATOGENIA
El desarrollo de las articulaciones se inicia en la quinta semana de vida embrionaria pudiendo apreciarse ya espacios articulares a la 7ª semana de gestación. Los movimientos del embrión comienzan a partir de la 8ª semana de vida. Es durante este primer trimestre donde se produce un desarrollo motor progresivo en todos los niveles, desde proliferación y migración de neuronas motoras hasta el desarrollo del tejido muscular.
El movimiento inicial del feto se genera por circuitos coordinados a nivel de la médula espinal y tronco, con un feedback que es mediado a través de las inmaduras fibras musculares de los miotomas. Los movimientos posteriores son secundarios a una coordinación supraespinal desde áreas cerebrales. El desarrollo de los movimientos en el feto sigue un patrón que se repite y es homogéneo en la especie humana, estando plenamente desarrollados ya a las 15 semanas de gestación. Los movimientos que aparecen primero son lo movimientos laterales del feto seguido de movimientos de sobresalto, de contracción, movimientos aislados de extremidades, movimientos respiratorios, hipo, movimientos de cabeza y cuello, así como movimientos de chupeteo, deglución, movimientos de la mandíbula, contacto manos-cara y movimientos de estiramiento y rotación 22. La detección del movimiento fetal por la madre comienza después de 16-18 semanas. Una disminución de los movimientos fetales después de la décima semana indica un mal desarrollo y/o disfunción de estructuras fetales neuromusculares centrales o periféricas. Tanto el movimiento embriónico como las contracciones musculares son esenciales en el normal desarrollo de las articulaciones, jugando ambas un rol esencial en la esqueletogénesis, existiendo una clara interdependencia entre estos procesos.
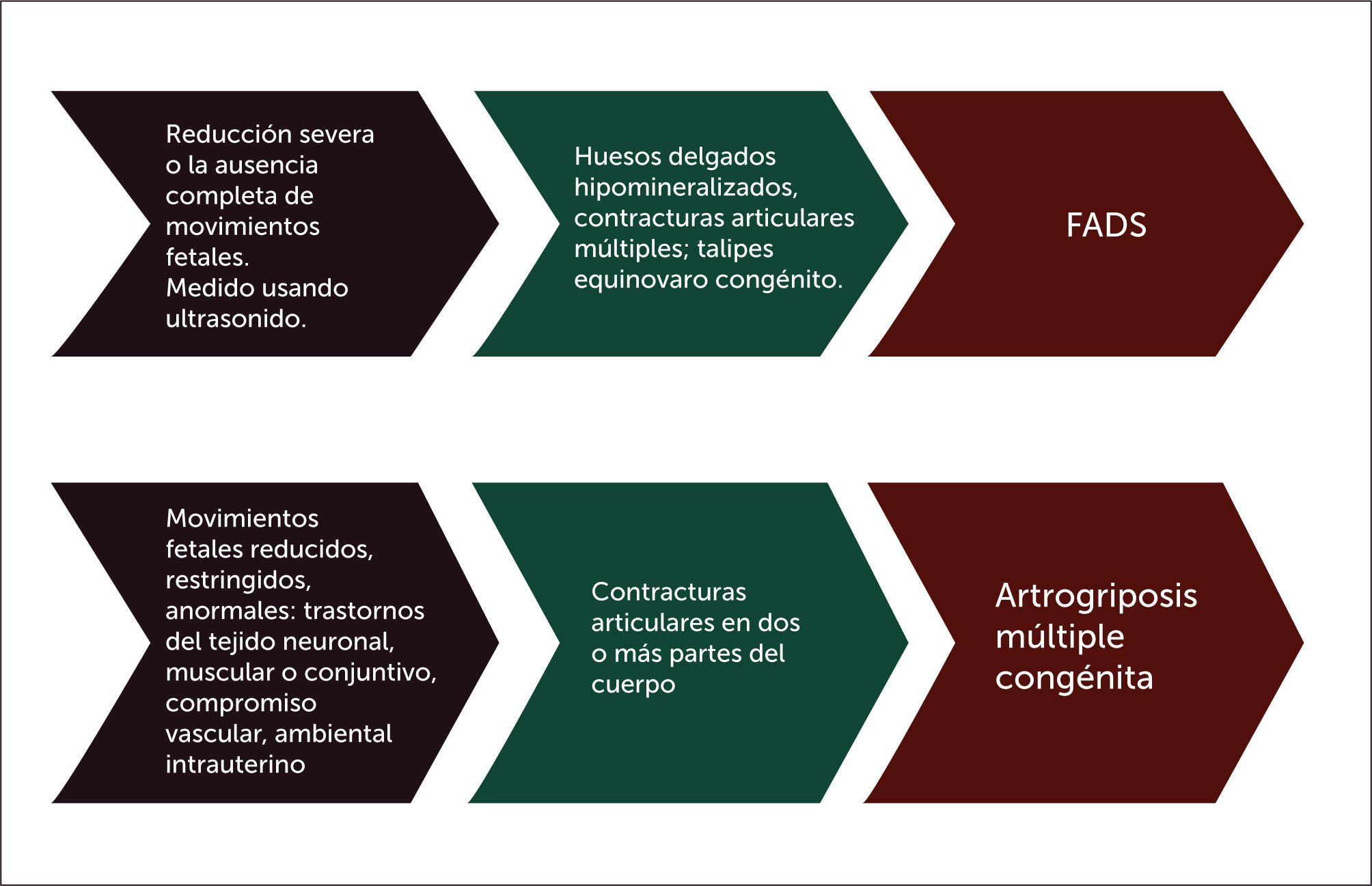
Es finalmente la restricción al normal movimiento in útero del feto, desde la ausencia prácticamente total que origina una secuencia de deformación de akinesia fetal (FADS), así como una alteración cualitativa y/o cuantitativa total o segmentaria, las que originarán una artrogriposis de severidad variable (Figura 3).
ETIOLOGÍA
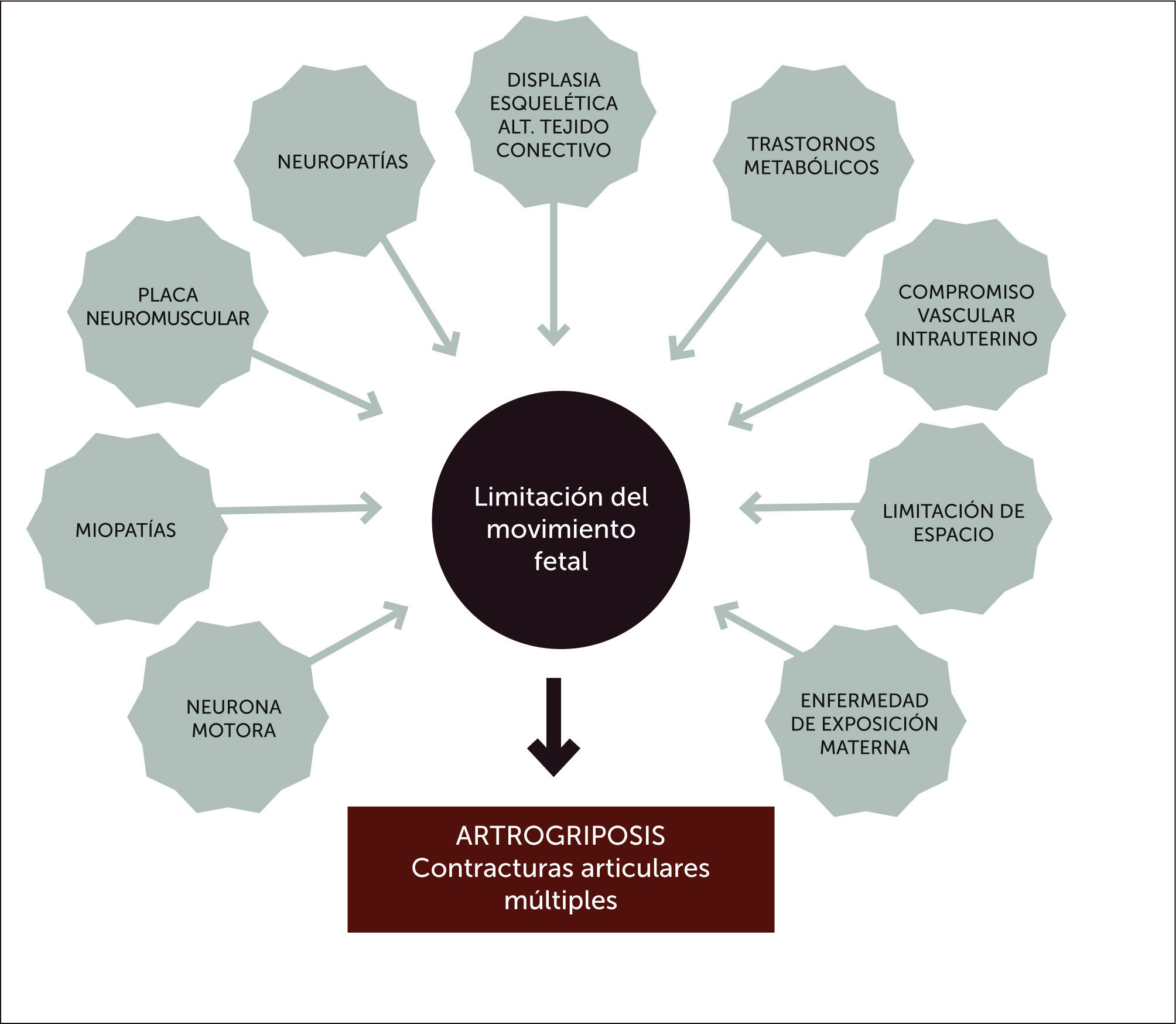
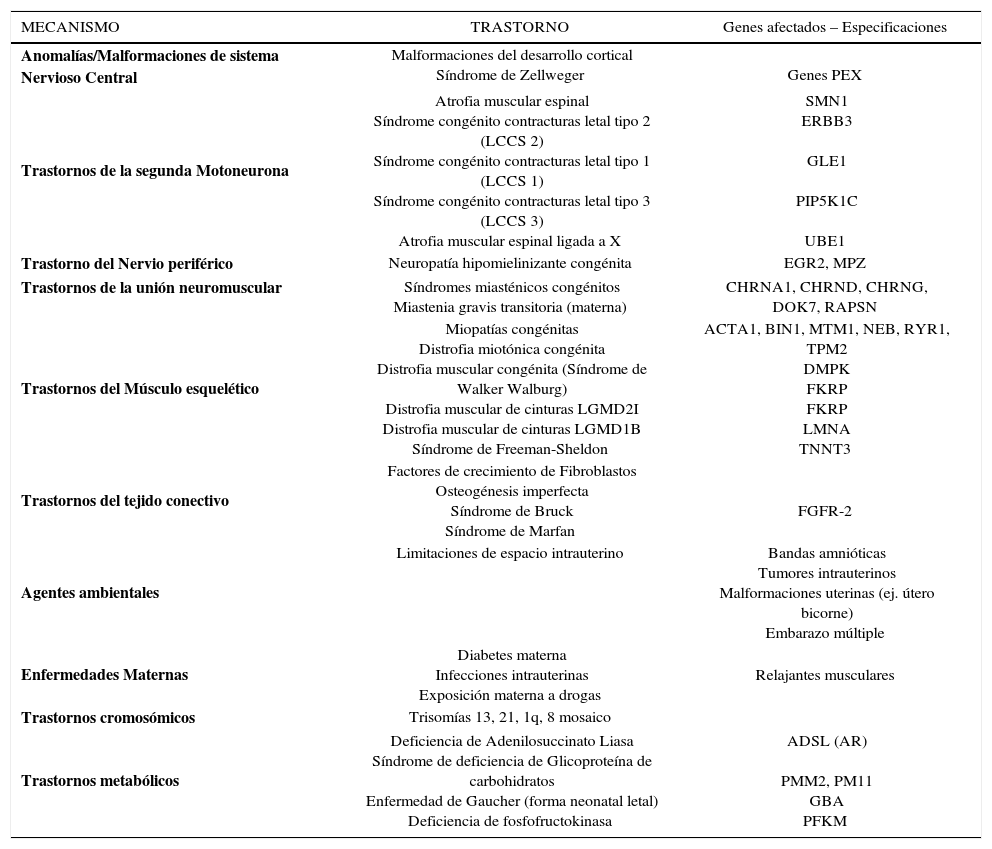
La akinesia/hipokinesia fetal puede ser causada por alteraciones en cualquier punto de las estructuras del sistema motor, desde el cerebro, médula espinal, nervio periférico, unión neuromuscular, músculo y tejido conectivo (Figura 4). En una revisión extensa sobre la etiología y clasificación de las AMC 1, se dividen a las posibles etiologías subyacentes en:

-Procesos miopáticos
-Procesos neuropáticos (incluyendo sistema nervioso central y periférico)
-Anormalidades de la placa neuromuscular
-Anormalidades del tejido conectivo
-Alteraciones cromosómicas
-Trastornos metabólicos
-Trastornos epigenéticos
-Limitaciones de espacio que conduce a una restricción del movimientos en el útero
-Enfermedad materna
-Exposición materna
-Compromiso del suministro de sangre a la placenta y/o embrión/feto
En la Tabla 3 se desglosan algunas de las principales etiologías y su causa genética específica.
CAUSAS DE AKINESIA/HIPOKINESIA FETAL
| MECANISMO | TRASTORNO | Genes afectados – Especificaciones |
|---|---|---|
| Anomalías/Malformaciones de sistema Nervioso Central | Malformaciones del desarrollo cortical Síndrome de Zellweger | Genes PEX |
| Trastornos de la segunda Motoneurona | Atrofia muscular espinal Síndrome congénito contracturas letal tipo 2 (LCCS 2) Síndrome congénito contracturas letal tipo 1 (LCCS 1) Síndrome congénito contracturas letal tipo 3 (LCCS 3) Atrofia muscular espinal ligada a X | SMN1 ERBB3 GLE1 PIP5K1C UBE1 |
| Trastorno del Nervio periférico | Neuropatía hipomielinizante congénita | EGR2, MPZ |
| Trastornos de la unión neuromuscular | Síndromes miasténicos congénitos Miastenia gravis transitoria (materna) | CHRNA1, CHRND, CHRNG, DOK7, RAPSN |
| Trastornos del Músculo esquelético | Miopatías congénitas Distrofia miotónica congénita Distrofia muscular congénita (Síndrome de Walker Walburg) Distrofia muscular de cinturas LGMD2I Distrofia muscular de cinturas LGMD1B Síndrome de Freeman-Sheldon | ACTA1, BIN1, MTM1, NEB, RYR1, TPM2 DMPK FKRP FKRP LMNA TNNT3 |
| Trastornos del tejido conectivo | Factores de crecimiento de Fibroblastos Osteogénesis imperfecta Síndrome de Bruck Síndrome de Marfan | FGFR-2 |
| Agentes ambientales | Limitaciones de espacio intrauterino | Bandas amnióticas Tumores intrauterinos Malformaciones uterinas (ej. útero bicorne) Embarazo múltiple |
| Enfermedades Maternas | Diabetes materna Infecciones intrauterinas Exposición materna a drogas | Relajantes musculares |
| Trastornos cromosómicos | Trisomías 13, 21, 1q, 8 mosaico | |
| Trastornos metabólicos | Deficiencia de Adenilosuccinato Liasa Síndrome de deficiencia de Glicoproteína de carbohidratos Enfermedad de Gaucher (forma neonatal letal) Deficiencia de fosfofructokinasa | ADSL (AR) PMM2, PM11 GBA PFKM |
Como se comentó más arriba, en cerca de dos tercios de los casos de FADS no se logra realizar el diagnóstico prenatal. Una vez evaluado el recién nacido es posible alcanzar un diagnóstico específico en el período neonatal sólo en el 50% de los casos. La observación y evolución en el tiempo, la respuesta a la terapia, el nivel intelectual y el desarrollo psicomotor, son claves que llevarán a un diagnóstico específico que puede llegar a resolver la etiología responsable hasta en un 75% de los casos a los dos años de edad 1.
Distintos autores agrupan en forma diferente las distintas causas responsables de la AMC. Hall et al, describen tres subtipos de trastornos orientadores de la causa etiológica que involucran múltiples contracturas: aquellas que incluyen principalmente extremidades (ej.: amioplasia, artrogriposis distal tipos 1-9, síndrome de Poland, camptodactilia), contracturas con anomalías de otros sistemas (ej.: displasia campomiélica, displasia de Larsen, displasia Kniest, displasia metafisiaria, etc.) y contracturas de extremidades con compromiso del sistema nervioso (ej.: síndrome de pterigium múltiple letal, artrogriposis letal ligada X, síndrome de pterigium con labio leporino, etc.) 1.
Existen pistas clínicas específicas que permiten orientar el momento en el que se produjo la restricción de los movimientos fetales, como la ausencia o disminución de los pliegues de flexión de los dedos y las extremidades, la presencia de pterigium, la gravedad del retraso del crecimiento intrauterino y la presencia de osteoporosis. La ausencia de movimiento fetal en el primer trimestre origina la presencia de pterigium o bandas de piel en las articulación. Sin embargo, puesto que la formación de las extremidades y en particular las formaciones de las articulaciones requieren del movimiento, la ausencia de movimiento sostenido in útero debería comenzar después de 8 semanas de gestación para que originen pterigium articulares1,11. En el diagnóstico diferencial, la presencia de compromiso del sistema nervioso es crucial para identificar etiología de las contracturas secundarias a compromiso del SNC o periférico.
En relación con las causas neuromusculares, es importante señalar que la akinesia fetal y la AMC representan el espectro más grave de manifestación clínica de diversas de entidades conocidas como atrofias musculares espinales, miopatías congénitas, miastenias congénitas etc., donde en el perfil evolutivo existen pobres mejorías a través de las terapias 5.
En una serie de nuestros casos (no reportados) de la Unidad de Medicina Fetal del Hospital Gustavo Fricke, la mayor parte de las alteraciones ocurren en las extremidades inferiores como pie bot en concordancia con la prevalencia antes descrita para esta entidad. En uno de nuestros casos la secuencia de hipokinesia fetal se asoció a una gastrosquisis fetal, la cual es una malformación relacionada con isquemia de origen vascular de la pared abdominal.
Aproximación diagnóstico prenatalComo hemos revisado más arriba, el síndrome de akinesia fetal corresponde a un conjunto heterogéneo de alteraciones anatómicas caracterizadas por una combinación variable de hallazgos secundarias a la hipomotilidad fetal como: restricción del crecimiento intrauterino, artrogriposis y otras alteraciones anatómicas que incluyen la hipoplasia pulmonar, el labio fisurado y la criptorquidia. Se trata de un diagnóstico sindromático con múltiples opciones de diagnóstico diferencial.
En relación a la detección prenatal, dada la multiplicidad de hallazgos anatómicos posibles, el estudio a través de ecografía debe estar orientado a hallazgos directos o indirectos que evidencien una disminución de los movimientos fetales. Como signos indirectos se debe sospechar esta condición en un feto portador de una restricción del crecimiento fetal asociado a polihidroamnios y como signos directos, la presencia de alteraciones esqueléticas o posturales de las extremidades que pueden afectar las manos y/o los pies (ej.: pie bot, pie equinovaro), que suelen ser de carácter bilateral, pudiendo existir una disarmonía en su severidad.
En cuanto a signos indirectos de hipokinesia, la restricción de crecimiento fetal se define como un peso fetal estimado menor al percentil 10 para la edad gestacional según las tablas de recién nacidos, sin embargo, en este caso pudiera ser necesario estimar la magnitud de la restricción de crecimiento fetal por tablas ecográficas de biometría fetal (perímetro abdominal) y considerar la opción de una alteración del crecimiento intrauterino de origen placentario por las alteraciones circulatorias en el doppler de arterias uterinas.
La definición del aumento de líquido amniótico es más subjetiva, aunque se ha intentado objetivar mediante indicadores semicuantitativos como el índice de líquido amniótico (mediciones bolsillo en cada cuadrante abdominal) o por el bolsillo vertical máximo. En general, la disminución de la deglución fetal puede considerarse como un fenómeno progresivo y por ello el determinante común es el aumento exponencial de líquido amniótico con el avance de la gestación.
El diagnóstico prenatal de hipokinesia fetal suele ocurrir en la segunda mitad de la gestación, sin embargo, en los casos de mayor severidad puede manifestarse por akinesia fetal, que puede ser detectada por ecografía prenatal a las 12 semanas de gestación. Los hallazgos ecográficos posibles de pesquisar comprenden la falta de movimientos de las extremidades, postura anormal persistente de las extremidades, falta de movimientos faciales, polihidroamnios debido a la disminución de la deglución fetal, hipoplasia pulmonar, cordón umbilical corto debido a la disminución de los movimientos fetales, restricción del crecimiento intrauterino, aumento de la nuca translucidez, edema nucal o higroma quístico en el primer trimestre, y la hidropesía fetal23,24.
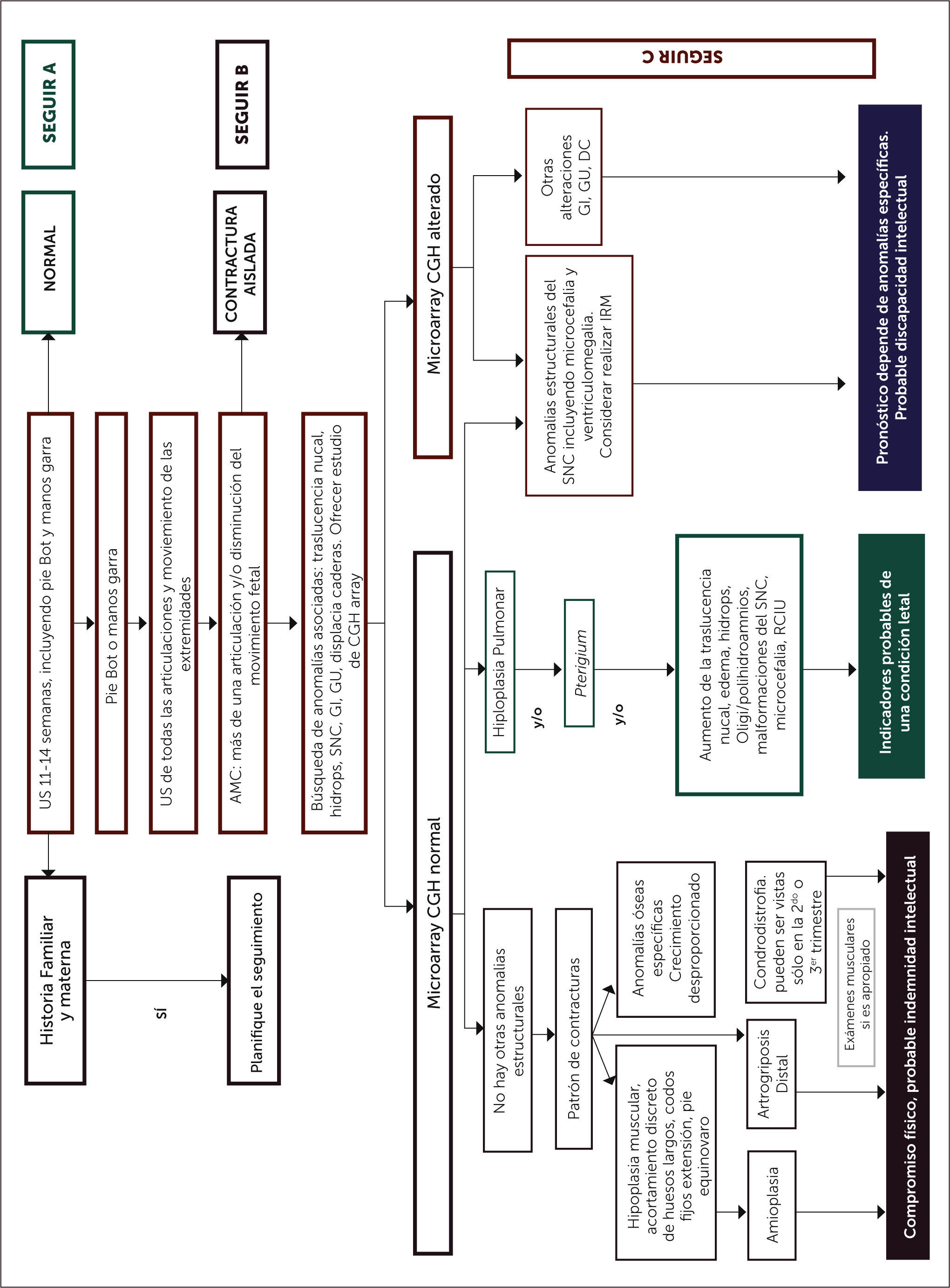
La ultrasonografía en tiempo real puede hacer un diagnóstico de estas condiciones, al visualizar la anatomía y el movimiento fetal, permitiendo detectar contracturas, posturas de articulaciones, la calidad de los movimientos fetales, mediciones pulmonares, tejido muscular y crecimiento óseo, tan precoz como en primer trimestre o al inicio del segundo semestre. El estudio propuesto por Filges et al 7, plantea una estrategia de diagnóstico prenatal que pensamos puede ser de gran utilidad para los clínicos enfrentados a esta patología (Figura 5). En dicho algoritmo se sugiere realizar US precoz entre las 11-14 semanas, con búsqueda dirigida de manos en garra y pie bot, análisis de la presencia de otras contracturas descartando posibilidad de contractura aislada, posterior revisión de compromiso de otros sistemas involucrados y diagnóstico diferencial con estudio genético y seguimiento por US seriado.
")
En la pesquisa diagnóstica prenatal de FADS o artrogriposis, especialmente en el tercer trimestre, puede ser útil la resonancia nuclear magnética fetal para la definición precisa de la anatomía cerebral y los movimientos del feto, con la desventaja que aún no es posible con esta técnica evaluar el movimiento en tiempo real. Los hallazgos neurológicos de una resonancia nuclear magnética pueden incluir agenesia de cuerpo calloso, lisencefalia, hidrocefalia o alteraciones de la médula espinal. Aunque la mayoría de estos hallazgos pueden evaluarse por neurosonografía cerebral, especialmente por vía transvaginal, la resonancia nuclear magnética tiene impacto significativo en el diagnóstico de alteraciones de la migración neuronal y de alteraciones de la línea media y fosa posterior. Algunos estudios han demostrado datos prometedores en cuanto a la capacidad de la resonancia fetal de detectar anomalías músculo-esqueléticas, movimiento fetal y desarrollo muscular 25.
Pese a los avances en la capacidad y resolución de las imágenes en medicina fetal existe aún una muy baja tasa de diagnóstico prenatal en las contracturas congénitas múltiples, con sólo el 22.2% casos diagnosticados prenatalmente 7, esto en gran medida porque no hay datos suficientes que permitan establecer normas sobre que se considera movimientos normales o anormales en las distintas etapas de la vida fetal. Como lo señalan otros autores, es muy importante establecer estándares de cuantificación de movimiento fetal de manera de poder pesquisar a tiempo alteraciones significativas del patrón esperado y facilitar un diagnóstico prenatal que podría por ejemplo permitir posibles intervenciones que mejoren el pronóstico del niño, tales como el aumento de los movimientos con cafeína o ejercicio materno1,7 o induciendo un parto prematuro.
Aproximación diagnóstico postnatalMás del 50% de las causas de AMC se deben a patología neuromuscular, con al menos 30 genes implicados y que afectan el adecuado funcionamiento de eje neuromuscular a todo nivel, desde motoneurona, nervio periférico, unión neuromuscular y tejido muscular. En un reciente estudio de casos de FADS, AMC y miopatías congénitas severas al nacer se pudo identificar en forma concluyente el 47% de la causa genética subyacente a través del estudio de exoma y de un panel de secuenciación de nueva generación (next generation sequencing) que incorporó todos los genes conocidos productores de enfermedades neuromusculares 6. Muchas miopatías congénitas y distrofias musculares pueden presentar al nacer contracturas articulares múltiples como algunas miopatías nemalínicas, centronucleares, miotubulares, etc. También se conoce que mutaciones en varios genes que se expresan en las fibras musculares de contracción rápida producen artrogriposis distales1,14,15. La presencia de pterigium múltiples debe hacer sospechar la posibilidad de mutaciones en genes relacionados con la placa neuromuscular como varias subunidades de los receptores de acetilcolina responsables del SE y síndrome de pterigium múltiple descritos anteriormente.
Junto a las patologías neuromusculares, un porcentaje importante de hipokinesias fetales y AMC de deben a malformaciones o compromisos del sistema nervioso central. Por este motivo el análisis inicial de estos pacientes debe considerar una extensa historia familiar de al menos 3 generaciones, antecedentes maternos y del embarazo así como un exhaustivo examen físico y neurológico del paciente. Frente a la sospecha de compromiso cognitivo o central se deben realizar neuroimágenes y la existencia de malformaciones de múltiples sistemas y dismorfias, realizar un estudio cromosómico. La Tabla 4 resume los puntos importantes que deben considerase en el estudio postnatal de estos pacientes y los exámenes de laboratorio que deben considerarse en el abordaje se indican en la Tabla 5.
ESTUDIO POSTNATAL
| EVALUACIÓN CLÍNICA | |
|---|---|
| 1. HISTORIA: EMBARAZO-PARTO HISTORIA FAMILIAR Toda condición que favorezca Hipomovilidad in útero | Miastenia gravis, distrofia miotónica, Diabetes en madre? Poli/oligo hidroamnios Fiebre >39°C, drogas: misoprostol, curare, alcohol; infecciones congénitas Otros familiares afectados? Hiperlaxitud familiar, Hipotonía, Miopatías, consanguinidad Parto en podálica, traumático, fracturas al nacer, masa uterina, estructura uterina |
| 2. EVALUACIÓN DEL RECIÉN NACIDO Descripción de contracturas y deformidades | Qué extremidades y articulaciones/Proximal vs distal/Flexión vs extensión Fusión completa o anquilosis vs contractura de tejido blando Limitación: fija, pasiva, movimiento activo/Posición característica en reposo Describir detalladamente todas las deformidades. Revisión por sistema Fascie, ojos, orejas, mandíbula, paladar, genitales, columna, piel, hoyuelos |
| 3. CURSO Y EVOLUCIÓN | Cambios con el tiempo, hitos del desarrollo, dificultades de alimentación, etc. |
| EXÁMENES DE LABORATORIO |
|---|
| FOTOGRAFÍAS/RADIOGRAFÍAS/ECOGRAFÍAS/ IRM CEREBRO-MÉDULA ESPINAL-MÚSCULOS Anomalías óseas, desproporción esquelética, escoliosis, anquilosis, luxación caderas, rótulas, cabeza radial etc. CARIOGRAMA/CGH ARRAY/DNA EXOMA Compromiso múltiples sistemas, sistema nervioso central EXAMEN GENÉTICOS ESPECÍFICOS SMN1/DMPK Atrofia muscular espinal/distrofia miotónica EMG/VCN Estimulación repetitiva biopsia muscular estudio mitocondriales Screening metabólico (organomegalia) cultivo fibroblastos Creatinkinasa total en plasma/Anticuerpos específicos: Anti Ach Cultivos virales, niveles de IgM en el RN Examen oftalmológico |
| AUTOPSIA |
EXÁMENES DE LABORATORIO EN PACIENTES CON ARTROGRIPOSIS MÚLTIPLE CONGÉNITA (AMC) AL NACER
| Estudio de ADN si fenotipo se ajusta a un trastorno conocido en el que existen pruebas genéticas disponibles Si se sospecha una atrofia muscular espinal realizar estudio gen SMN1 |
| Estudio de ADN si fenotipo se ajusta a un trastorno conocido en el que existen pruebas genéticas disponibles Estudios de exoma si familia disponible |
| Sospecha infecciones congénitas: Cultivo viral, anticuerpos específicos, niveles de IgM en el RN |
| Creatinkinasa: si hay debilidad generalizada, masa muscular disminuida, Si CPK está elevada hacer biopsia muscular EMG-VCN: si sospecha neuropatía, motoneurona |
| Biopsia muscular: en zonas normales y afectadas en el momento de cirugías para distinguir compromiso miopático Examen oftalmológico: opacidades, degeneración de la retina, etc. |
| Anticuerpos materno para neurotransmisores (miastenia gravis) |
| Estudio de ADN mitocondrial si se sospecha mitocondriopatía |
| Screening metabólico si existe organomegalia |
El principal objetivo del tratamiento es lograr una movilización lo más precoz posible de las articulaciones para mejorar la función y posición de éstas y preservar los músculos de la atrofia por desuso. La terapia física, ocupacional, las férulas y los yesos seriados son parte importante de la terapia de todos los pacientes. Se debe tener especial cuidado con la terapia física para evitar fracturas iatrogénicas de los huesos largos ya que no debemos olvidar que a menudo son huesos osteoporóticos. Los pacientes con AMC son niños que recibirán por años apoyo multidisciplinario y terapia física de rehabilitación. Aquellas deformaciones de las extremidades persistentes que limitan la función deberán ser tratadas quirúrgicamente.
Los principales objetivos a largo plazo del tratamiento son finalmente aumentar la movilidad articular, la fuerza muscular y el desarrollo de patrones funcionales que permitan alcanzar la mayor independencia posible en las actividades de la vida diaria.
SÍNTESISLa akinesia/hipokinesia fetal incluye un espectro amplio de trastornos que asociados a la disminución o ausencia de movimientos fetales. Con los avances alcanzados en las últimas décadas en este trastorno, con la identificación de múltiples enfermedades y cientos de alteraciones genéticas asociadas, es un desafío establecer la etiología subyacente en cada caso particular. Ha sido, sin duda, el desarrollo de la genética molecular, la principal herramienta de avance en estos trastornos en los últimos años.
Patologías a cualquier nivel de las estructuras del sistema motor, desde el cerebro hasta el músculo y tejido conectivo, pueden ser causa de síndromes de akinesia/hipokinesia fetal, lo que plantea un amplio espectro diagnóstico. El difícil proceso del diagnóstico diferencial y la búsqueda del diagnóstico etiológico debe considerar que un importante porcentaje de trastornos son explicados por patologías neuromusculares y malformaciones o compromiso del sistema nervioso central.
Avances en la detección prenatal han sido también muy significativos, dado el aporte de los estudios ecográficos convencionales, con hallazgos directos o indirectos de hipomovilidad fetal; el aporte de la ultrasonografía en tiempo real, en lo que respecta a la calidad del movimiento fetal; y los avances en estudios genéticos y en resonancia nuclear magnética fetal, entregando esta última información precisa de la anatomía cerebral y los movimientos del feto. A pesar de todos estos progresos, aún existe una muy baja tasa de diagnóstico etiológico prenatal en las contracturas congénitas múltiples.
La no existencia de un tratamiento específico no implica la ausencia de terapias en este grupo de trastornos, las que deben estar dirigidas hacia una movilización precoz y la mejoría de la función y posición articular, buscando evitar la atrofia muscular por desuso, y alcanzar así, en el largo plazo, la mayor independencia y funcionalidad en la vida diaria.
Los autores declaran no tener conflictos de interés, en relación a este artículo.