




En esta revisión se analizan las características hereditarias de las principales condiciones neurodegenerativas (demencias tipo Alzheimer y frontotemporal, enfermedad de Parkinson y otros cuadros como la enfermedad de Huntington). Se hace énfasis en la necesidad de contar con exámenes genéticos de confirmación diagnóstica en estos cuadros, lo que constituye una falencia en nuestro medio. Disponer de estos recursos diagnósticos permite una mejor aproximación al paciente, estableciendo un diagnóstico más certero, un pronóstico con más base y una asesoría genética adecuada. La implementación de una terapia efectiva para cada cuadro, estará basada en un futuro próximo en un diagnóstico más preciso de la condición del paciente y para esto los estudios genéticos son una herramienta muy importante de la que dispone el clínico, además de estudios de imágenes y de laboratorio con otros marcadores humorales.
In the present article we review the inheritance of relevant neurodegenerative disorders (Alzheimer's disease, fronto-temporal dementia, Parkinson‘s disease and Huntington‘s disease). We highlight the relevance of molecular diagnosis technologies, which are currently unavailable in our country. These resources allow providing better patient care with a more reliable diagnosis and prognosis, as well as an opportunity for genetic counseling. In the near future, therapies will be focused on specific disease mechanisms, and therefore accurate diagnosis will be essential.
La mayoría de los casos de demencias, incluyendo la enfermedad de Alzheimer (EA) y la demencia fronto-temporal (DFT), así como de síndromes parkinsonianos se presentan en personas sin antecedentes familiares, es decir, son casos esporádicos, cuya causa es multifactorial y poligénica. Por otra parte, los pacientes que tienen antecedentes en familiares de primer grado, o que debutan con síntomas en edades tempranas corresponden a una forma de presentación de cuadros neurodegenerativos que orienta hacia un síndrome de causa genética.
Otros trastornos neurodegenerativos que tienen una herencia monogénica son la enfermedad de Huntington (EH), ataxias cerebelosas autosómico dominantes y recesivas, otros coreas como neuroacantocitosis y neuroferritinopatías, entre muchos otros cuadros neurológicos.
En los cuadros de inicio temprano, el deterioro cognitivo puede ser el síntoma cardinal o asociarse a otras manifestaciones clínicas prominentes, tales como trastornos del movimiento, manifestaciones psiquiátricas, ataxia, enfermedad de motoneurona o migraña. De cualquier forma, debido al menor índice de sospecha en individuos jóvenes, existe una mayor probabilidad de atribuir erróneamente las manifestaciones a un trastorno mental, retrasando el diagnóstico.
Las manifestaciones clínicas de estas enfermedades se superponen, por lo que la anamnesis, examen físico y exámenes de apoyo no bastan para establecer el diagnóstico definitivo. Ello requeriría la identificación de mutaciones causales en el afectado o portador. El diagnóstico molecular en etapas tempranas o presintomáticas tiene muchas aplicaciones relevantes 1.
Si bien no existen intervenciones que permitan prevenir la aparición de una de estas enfermedades o curarlas, el diagnóstico preciso favorece una mejor planificación de los cuidados que el afectado requerirá. Esto es de gran relevancia para la calidad de vida de las familias.
Por otra parte, existe una tendencia creciente entre familiares de pacientes con enfermedades neurodegenerativas a interesarse por identificar su estatus de portadores. Ello les permitiría tomar decisiones reproductivas, implementar medidas que permitan retrasar la aparición de los síntomas y establecer un diagnóstico precoz. Se ha establecido que contar con el diagnóstico de una enfermedad neurodegenerativa heredable tiene beneficios emocionales al reducir la ansiedad asociada a la incertidumbre y al proveer información necesaria para la toma de decisiones 2.
Los ensayos clínicos para fármacos que pudieran beneficiar a los pacientes requieren la evaluación del efecto en una muestra de pacientes con una misma condición, ya que los mecanismos fisiopatológicos del deterioro son diferentes entre ellas. Actualmente están en evaluación terapias gen-específicas por lo que el diagnóstico preciso cobrará cada vez más importancia práctica 3. La identificación de portadores es relevante para el desarrollo de ensayos clínicos en etapas en las que el daño celular propio de las enfermedades aun no está instalado. Por ejemplo, en el último año se inició la administración de potenciales modificadores de la expresión de la huntingtina mutante (proteína alterada en EH) en individuos portadores asintomáticos de la mutación y quienes tarde o temprano desarrollarían la -hasta ahora letal- enfermedad.
A nivel de investigación básica, los hallazgos en pacientes con formas familiares de estas enfermedades han contribuido a expandir el conocimiento de los mecanismos involucrados en las formas esporádicas. Es el caso de la identificación del rol de synucleina y LRRK2 en la enfermedad de Parkinson (EP).
En la actualidad, se encuentran disponibles en el mercado exámenes para identificar mutaciones causales de las patologías de interés. Ellos se basan en técnicas de secuenciación de última generación que pueden enfocarse en el análisis de un gen (por ejemplo, para el diagnóstico de EH), en un grupo de genes asociados a una misma enfermedad (por ejemplo, EA, DFT), o a un grupo amplio de enfermedades (por ejemplo, panel de demencias por medio de secuenciación de última generación). La secuenciación masiva, con cobertura de exoma completo o genoma completo, suele no estar indicada debido a que la mayoría de los casos son explicados por algunas de las mutaciones cubiertas por los paneles disponibles, y por el alto número de hallazgos inespecíficos que pueden surgir de este tipo de estudios 1. El tipo de examen a solicitar depende del tipo de síntomas del paciente y eventuales familiares afectados y del número de genes conocidos para los diagnósticos presuntivos.
En Chile en la actualidad el diagnóstico molecular de algunas enfermedades monogénicas se realiza para casos individuales. Si bien las condiciones estudiadas con mayor interés son los trastornos del desarrollo 4, también se han reportado algunos casos de enfermedades neurodegenerativas en que se ha confirmado la sospecha diagnóstica 5–9. Sin embargo, la disponibilidad de centros y profesionales para efectuar la asesoría genética, así como para realizar las técnicas de biología molecular, el análisis bioinformático y la interpretación de los resultados, está lejos de cumplir las necesidades de la población chilena 10. El contar con estos servicios en nuestro medio mejoraría la atención ofrecida a los pacientes y familias de nuestro país. Es esperable que, al menos en una etapa inicial, los costos de las técnicas de biología molecular sean mayores que en el extranjero debido a que dichas técnicas han sido optimizadas para grandes cantidades de muestras. Sin embargo, habría una ventaja clara en cuanto a los riesgos de pérdida de trazabilidad de la muestra o de brechas en la confidencialidad de los datos. Estos estudios no están cubiertos por las aseguradoras de salud, y para enfrentar esta brecha en el acceso a los servicios es imprescindible que sean realizados en nuestro país.
Más fundamental aún, desarrollar localmente estas herramientas permitiría efectuar el proceso diagnóstico en el contexto adecuado de trabajo en equipo incluyendo al médico tratante y al profesional que realice el asesoramiento genético. El médico tratante es quien plantea un diagnóstico presuntivo de manera de establecer la correcta indicación del examen de diagnóstico molecular. El asesoramiento genético debe efectuarse en la etapa previa y posterior a la realización del examen, incluyendo información completa sobre cuál es el alcance del estudio, la adecuada interpretación de los resultados y asesoría en relación a la eventual inclusión de familiares cuyo riesgo pueda concluirse de este primer estudio, considerando siempre el derecho a no querer saber. Sin estos elementos, la revelación de un diagnóstico desfavorable, puede resultar en un malestar emocional incluso severo 11.
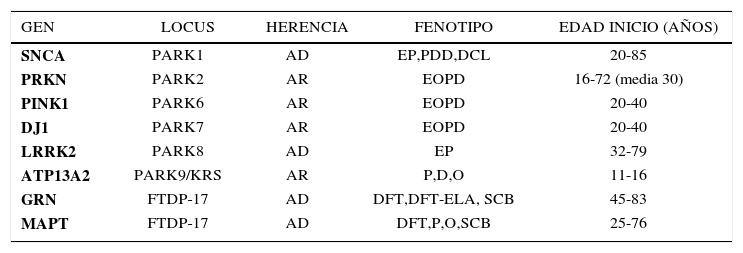
GENÉTICA DE LAS ENFERMEDADES NEURODEGENERATIVAS HEREDITARIAS ENFERMEDAD DE PARKINSON (VER TABLA 1)a) Formas autosómico dominantesUn hito importante en la historia de esta enfermedad se produjo en 1997 cuando se comunicó el estudio de la familia Contursi, llamada así por la localidad en Italia donde residía, con más de 50 afectados en 5 generaciones 12. Esta familia presentó una forma autosómica dominante (AD) de EP que por otra parte era bastante similar en los aspectos clínicos a la forma esporádica y clásica descrita por James Parkinson en 1817. El estudio genético de la familia señalada llevó al descubrimiento del rol de la proteína synucleína en la patogenia de la enfermedad 13. La synucleína está presente en los cuerpos de Lewy, inclusión intraneuronal característica a nivel patológico de la enfermedad. El gen SNCA que codifica la proteína synucleína presentaba duplicaciones o triplicaciones: a mayor carga de la mutación, más severo el fenotipo, semejando a la demencia por cuerpos de Lewy o atrofia multisistémica. El rol de esta proteína sináptica neuronal no está aclarado, pero se sabe que es importante en la transmisión de información interneuronal. Se observa esta mutación hasta en el 2% de los casos familiares con herencia AD 14. Más frecuente es la mutación del gen LRRK2 (acrónimo de leucine-richrepeatkinase 2) que da cuenta de 10-20% de los casos AD 7, y también se observa en casos esporádicos. La clínica también es indistinguible de la forma clásica de Parkinson. La penetrancia de la expresión clínica es edad dependiente y varía de 30 a 70% a los 80 años 15.
GENES IMPLICADOS EN FORMAS MONOGENÉTICAS DE LA ENFERMEDAD DE PARKINSON Y PARKINSON ATÍPICOS
| GEN | LOCUS | HERENCIA | FENOTIPO | EDAD INICIO (AÑOS) |
|---|---|---|---|---|
| SNCA | PARK1 | AD | EP,PDD,DCL | 20-85 |
| PRKN | PARK2 | AR | EOPD | 16-72 (media 30) |
| PINK1 | PARK6 | AR | EOPD | 20-40 |
| DJ1 | PARK7 | AR | EOPD | 20-40 |
| LRRK2 | PARK8 | AD | EP | 32-79 |
| ATP13A2 | PARK9/KRS | AR | P,D,O | 11-16 |
| GRN | FTDP-17 | AD | DFT,DFT-ELA, SCB | 45-83 |
| MAPT | FTDP-17 | AD | DFT,P,O,SCB | 25-76 |
Abreviaturas: AD, autosomico dominante; AR, autosomico recesivo; SCB, síndrome cortico-basal; D, demencia; DCL, demencia con Cuerpos de Lewy; EOPD, Enfermedad de Parkinson de inicio precoz; DFT, demencia frontotemporal; DFT-ELA, demencia frontotemporal-enfermedad de motoneurona; DFTP-17, demencia frontotemporal con parkinsonismo ligada al cromosoma 17; SKR, Sìndrome de Kufor-Rakeb; O, signos oculomotores; P, parkinsonismo; EP, Enfermedad de Parkinson; PDD, Demencia asociada a la enfermedad de Parkinson.
El hallazgo de mutaciones en el gen SNCA abrió el paso a la investigación de la importancia de una serie de genes cuya mutación explica también formas autosómico recesivas (AR) de la enfermedad, con una edad menor de presentación, en general antes de los 40 años. Se han descrito mutaciones homocigotas y heterocigotas compuestas en 3 genes principales: PRKN (parkin, causante de la forma denominada PARK2), PINK1 (causante de PARK6), y DJ-1 (causante de PARK7) 16,17. Las mutaciones en PRKN son las más comunes y explican la mitad de los casos familiares compatibles con una herencia AR, con una edad de inicio antes de los 45 años, y también de un 15% de los casos esporádicos con inicio antes de los 45 años. Las otras mutaciones en PINK1 y PARK7 son más infrecuentes, dando cuenta de hasta 8% de los casos familiares y 1% a 2% de los esporádicos de inicio precoz, respectivamente. La posibilidad de la presencia de mutaciones en estos genes es función de la edad de inicio: mientras más precoz, más alta la probabilidad de hallar mutaciones. Existe información de la patología subyacente en algunos pacientes con mutación en PRKN en quienes no se observan los típicos cuerpos de Lewy, lo que sugiere que estas formas son distintas en su patogenia de las formas AD. El fenotipo asociado a las mutaciones en PRKN se caracteriza por un parkinsonismo de inicio precoz, respuesta muy buena y prolongada a levodopa o agonistas, y a menudo un curso benigno. El promedio de edad de inicio es alrededor de los 30 años en la mayoría de los pacientes, pero también se han descrito casos más tardíos. Las fluctuaciones motoras y disquinesias por levodopa son frecuentes, mientras que es raro que haya alteraciones cognitivas o autonómicas. El fenotipo de mutaciones PINK1 y DJ-1 es muy similar a lo descrito para PRKN. Además mutaciones en otros 3 genes, ATP13A2 9, PLA2G6 y FBXO7, causan formas más raras de parkinsonismo recesivo (PARK9, PARK14 y PARK15, respectivamente). Ellas se caracterizan por un inicio muy precoz (<30 años) y características clínicas atípicas (signos piramidales, distonía, alteraciones oculomotoras y cognitivas) 18.
ENFERMEDAD DE ALZHEIMERSe diagnostica la forma familiar de EA en familias que tienen más de un miembro con demencia (usualmente varias personas afectadas en más de una generación) en la cual la edad de inicio de la enfermedad es consistentemente antes de los 60-65 años y a menudo antes de los 55 años.
Se conocen principalmente mutaciones en 3 genes en esta variedad de Alzheimer (ver Tabla 2):
GENES ASOCIADOS CON DEMENCIAS HEREDITARIAS
| GEN | ENFERMEDAD | PENETRANCIA A LO LARGO DE LA VIDA (%) |
|---|---|---|
| APP | EA | 100 |
| PSEN1 | EA | 100 |
| PSEN2 | EA | Cercano a 100 |
| APOE | EA | FACTOR DE RIESGO (disponible CLC) |
| MAPT | DFT | Cercano a 100 |
| GRN | DFT | Cercano a 100 |
| C9orf72 | DFT-ELA | Cercano a 100 |
| FUS | DFT-ELA | Cercano a 100 |
| HTT | EH | 100 |
| PRNP | ECJ | Cercano a 100 |
(Adaptado de Goldman, 2015).
Abreviaturas: EA: Enfermedad de Alzheimer; DFT: Demencia fronto-temporal; DFT-ELA: Demencia fronto-temporal y enfermedad de motoneurona; EH: Enfermedad de Huntington; ECJ: Enfermedad de Creutzfeldt–Jakob.
PSEN1 (Presenilina 1), mutaciones en este gen dan cuenta de 30%-70% de todos los casos precoces 19; APP (Proteína precursor del amiloide), una de las primeras mutaciones descritas y constituyen entre el 10-15% de los casos; y PSEN2 (Presenilina 2) que dan cuenta de no más del 5% de las formas familiares 20.
DEMENCIA FRONTOTEMPORALEn una demencia de aparición precoz (< de 55 años) también es importante descartar una DFT, que según las actuales cifras de prevalencia constituye la demencia degenerativa de aparición precoz más frecuente (ver en este mismo número los artículos de Drs. Lillo y Leyton). Una de las mutaciones más reconocidas en la actualidad corresponde a la expansión de una secuencia repetida de seis nucleótidos en el gen C9orf72 21. Esta mutación es especialmente frecuente cuando existe historia familiar de enfermedad de motoneurona, pero también se encuentra en casos sin este antecedente.
ENFERMEDAD DE HUNTINGTON (EH)La EH se caracteriza por corea, deterioro cognitivo y ataxia. Se transmite en forma AD con un 100% de penetrancia. Constituye la principal causa a descartar en un adulto con corea, aun sin historia familiar clara. Su prevalencia mundial es de 5 a 10 por 100.000 habitantes.
La EH es la primera enfermedad neurológica en que pudo identificarse el producto de una mutación genética que se localizó en el cromosoma 4 en 1983 y luego el año 93 pudo identificarse el gen afectado. Esta investigación pionera se realizó en una comunidad alrededor del Lago Maracaibo en Venezuela, donde existe una alta frecuencia (asociada a un fenómeno evolucionario conocido como “efecto fundador”), llegando a 700 por 100.000 habitantes.
En la EH el defecto genético está en el gen HTT que codifica la proteína huntingtina y consiste en la expansión del trinucleótido CAG que codifica el aminoácido glutamina. La proteína codificada por el gen mutado presenta una interacción anormal con otras proteínas que lleva a muerte neuronal. Un individuo que tiene el gen HTT con 40 o más repeticiones de CAG va a desarrollar la enfermedad en algún momento de su vida. Sus hijos tienen 50% de riesgo de heredar la expansión de CAG y desarrollar esta enfermedad. La expansión de 35 a 39 repeticiones tiene una penetrancia incompleta. Además, presenta inestabilidad durante la gametogénesis de manera que puede dar origen a gametos portadores de una expansión patogénica y así producir casos de la enfermedad en la descendencia. Esto último se aplica también a la expansión de entre 25 y 34 repeticiones 22.
CONCLUSIONESEn la actualidad existe un amplio conocimiento sobre las bases moleculares de las formas hereditarias de enfermedades neurodegenerativas. Gracias a ello ha sido posible diseñar exámenes que permiten identificar mutaciones específicas que confirman el diagnóstico en un individuo afectado, o el riesgo para un portador. Si bien el diagnóstico genético en las enfermedades neurodegenerativas no se traduce aún en tratamientos específicos, sí contribuye a mejorar la calidad de vida del paciente y su familia. Futuros tratamientos que apunten a mecanismos patogénicos específicos están actualmente en investigación.
Sin duda existe una deuda con nuestros pacientes que sufren de estas distintas condiciones neurológicas. La medicina en Chile se ha quedado atrás en implementar importantes recursos necesarios para que el clínico confirme estos diagnósticos, ofrezca un pronóstico y consejo al paciente y su familia. Para que esto sea realidad, debe contarse con capacidades tecnológicas adecuadas pero además con las capacidades profesionales necesarias para un proceso diagnóstico exitoso. La indicación de un estudio molecular requiere de la evaluación por parte de clínicos expertos con un alto nivel de sospecha y con un conocimiento actualizado de las estrategias de estudio. El asesoramiento genético es determinante para que esta intervención resulte efectivamente en beneficio para el paciente y sus familiares. Al mismo tiempo, la difusión de estas condiciones hacia la población general permitirá una mejor interacción de los posibles afectados con los profesionales de la salud. La preocupación de nuestro sistema de salud por estos problemas se refleja en que ya existe un plan de salud de apoyo a la EP y se está implementando un Plan Nacional para las Demencias. Fomentar el acceso a servicios genéticos capaces de responder a las necesidades de los afectados por estas condiciones es un desafío para nuestro país.
Los autores declaran no tener conflictos de interés, en relación a este artículo.