





La enfermedad inflamatoria intestinal (EII) es una patología de etiología multifactorial y compleja, con aspectos conocidos y otros en investigación. Si bien tanto la Enfermedad de Crohn (EC) como la Colitis Ulcerosa (CU) han sido consideradas idiopáticas, se han identificado factores genéticos y ambientales que predisponen a estas enfermedades. Dependiendo del subtipo de la enfermedad, la inflamación puede manifestarse a nivel de la mucosa del colon en el caso de la CU, o bien, ser transmural y pudiendo comprometer cualquier parte del tubo digestivo en el caso de la EC. El proceso inflamatorio además afecta otros órganos fuera del tubo digestivo, como la piel, ojos, hígado, ductos biliares y articulaciones. La participación del sistema inmune es central, existiendo un equilibrio entre respuestas inmunes innatas y adaptativas, con complejas interacciones con la microbiota intestinal, así como otros factores ambientales. En la EII, este equilibrio se rompe, lo que conduce a una inflamación intestinal descontrolada. El conocimiento progresivo de la etiopatogenia ha contribuido a producir terapias dirigidas a bloquear procesos centrales de la EII que han permitido disminuir la terapia inmunosupresora convencional, reemplazándola por agentes biológicos seguros y precisos. En la presente revisión analizaremos distintos factores que determinan e influyen en la fisiopatología y expresión de la EII, estos incluyen factores hereditarios, de regulación inmunológica y los ambientales. Además, revisaremos cómo estos descubrimientos están relacionados al desarrollo de terapia cada vez más específica.
Inflammatory Bowel Disease (IBD) is a pathology of a complex and multifactorial etiology, with both known aspects and others in investigation. Although Crohn's Disease (CD) and Ulcerative Colitis (UC) are considered idiopathic, both genetic and environmental factors have been identified that predispose to these diseases. Depending on the subtype, inflammation may manifest at the colon mucous membrane as in UC, or can be transmural and it may compromise any segment of the digestive tube as in CD. The inflammatory process may extend to extraintestinal areas such as skin, eyes, liver, bile ducts and joints.
Immune system participation is key to this process, with a balance between immune and adaptative responses involving complex interactions with the gut flora, as well as others environmental factors. In IBD, this balance is broken, leading to uncontrolled intestinal inflammation. Progressive knowledge of the etiopathogenesis has contributed to develop therapies that aim towards blocking central processes of IBD that allow to lower the conventional immunosuppressive treatment, replacing it with secure and precise biological agents. In this article, we will analyze several factors that determine and influence the physiopathology and expression of the IBD, including hereditary, immune regulation and environmental factors. We will also review
how these findings are related to the development of physiopathology-directed therapy.
La fisiopatología de la enfermedad inflamatoria intestinal (EII) es compleja. Se ha demostrado que existen numerosos factores que interfieren en la etiología y patogenia tanto de Enfermedad de Crohn (EC) como en Colitis Ulcerosa (CU). Hay una influencia genética, del sistema immune y un componente ambiental que desarrollaremos a continución (fig. 1).
2Influencia genética
Se ha visto que la EII resulta de muchos factores que finalmente influencian a un huésped genéticamente susceptible. En estudios de gemelos, se ha observado una concordancia del 40 a 50% de EC, incluso en gemelos monocigotos. De este hecho se derivan dos observaciones: los factores ambientales siguen siendo determinantes en la patogenia de la EII, pero por otro lado los factores genéticos tienen un rol importante en el inicio de la enfermedad. Posteriormente se descubrió que el patrón de herencia no es Mendeliana simple sino un desorden genético complejo poligénico.
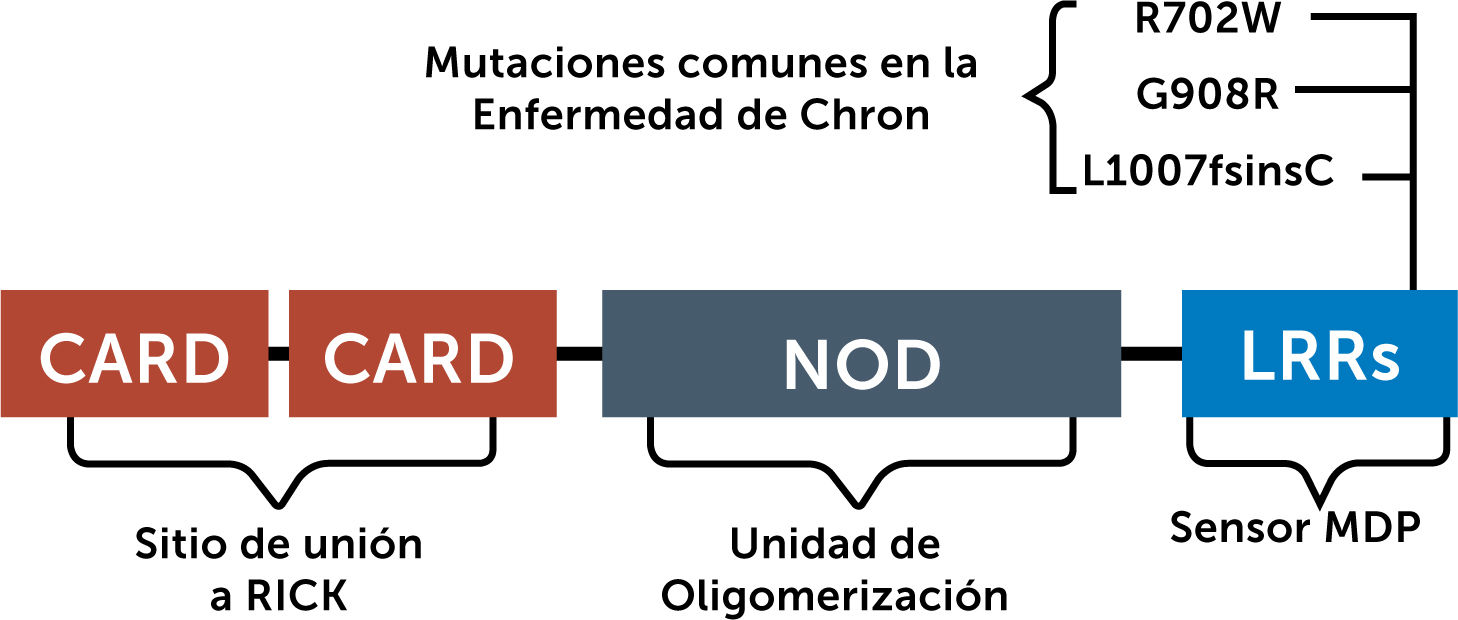
1.1 Nucleotide oligomerization domain (NOD): El año 2001 se describe la primera región de asociación genética de la EII, localizada en el cromosoma 161. En este cromosoma se encuentra el gen NOD2 (nucleotide oligomerization domain 2), con 3 polimorfismos posibles2. El ser heterocigoto confiere un riesgo modesto de EC (2 a 4 veces el riesgo), pero 2 o más alelos confieren un riesgo de 20 a 40 veces. NOD2 es un receptor de reconocimiento de patrones, expresado principalmente en células del sistema inmune innato, así como en células epiteliales intestinales. Se une a dipéptidos muramil, que son componentes de la pared celular bacteriana. Las variantes de NOD2 que se asocian a una susceptibilidad incrementada al desarrollo de EC muestran una función de reconocimiento deficiente de estos productos bacterianos, apuntando a la importancia de la microbiota en la patogenia de la EII. La estructura del NOD2 y sus mutaciones más frecuentes se muestran en la figura 2:

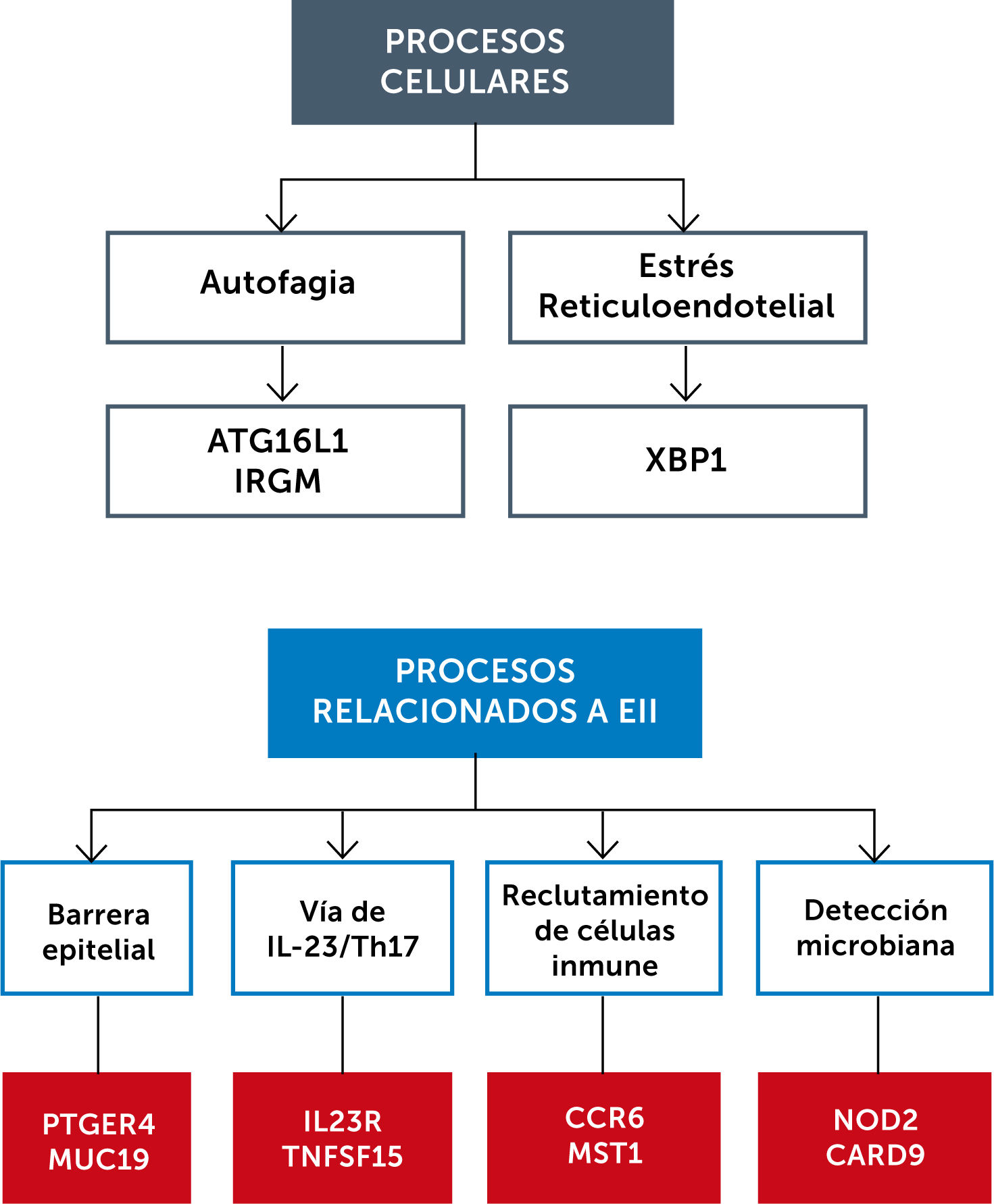
1.2 Genome-wide associatio, study (GWAS). Posteriormente surgen los GWAS (Genome-wide association study; del español estudios de asociación del genoma completo) para identificar múltiples genes que tienen menos peso que NOD2. Se han hecho 15 estudios GWAS en EII y se han identificado más de 250 locus de enfermedad3. La mayoría de los genes compartidos entre CU y EC tienen la misma dirección de efecto: las variantes de expresión génica exacerban o protegen de igual forma, lo que sugiere una función similar en ambas enfermedades.
Otro hallazgo interesante es que hay muchos locus que se comparten con otras enfermedades inflamatorias como psoriasis y espondilitis anquilosante4.
Además, se han descubierto varias vías biológicas que explican el desarrollo de la EII. La figura 3 muestra algunas de estas vías.

Pese a que los GWAS han identificado esta gran cantidad de locus asociados a EII, estas variantes genéticas sólo contribuyen aproximadamente a una heredabilidad del 26% en la EC y a un 19% en CU. En 2017, un grupo multicéntrico logró organizar los genes en redes de regulación inflamatoria, identificando grupos de genes claves que intervienen como iniciadores en la patogenia de la enfermedad5.
3Defectos inmunesUn elemento central en la patogenia de la EII es una función aberrante del sistema inmune de mucosas. Se han identificado defectos en el sistema inmune innato y adaptativo.
2.1 El sistema inmune innato consta de las células epiteliales de barrera, neutrófilos fagocíticos y macrófagos, las células NK y sus mediadores que colectivamente responden a antígenos tóxicos y/o patológicos.
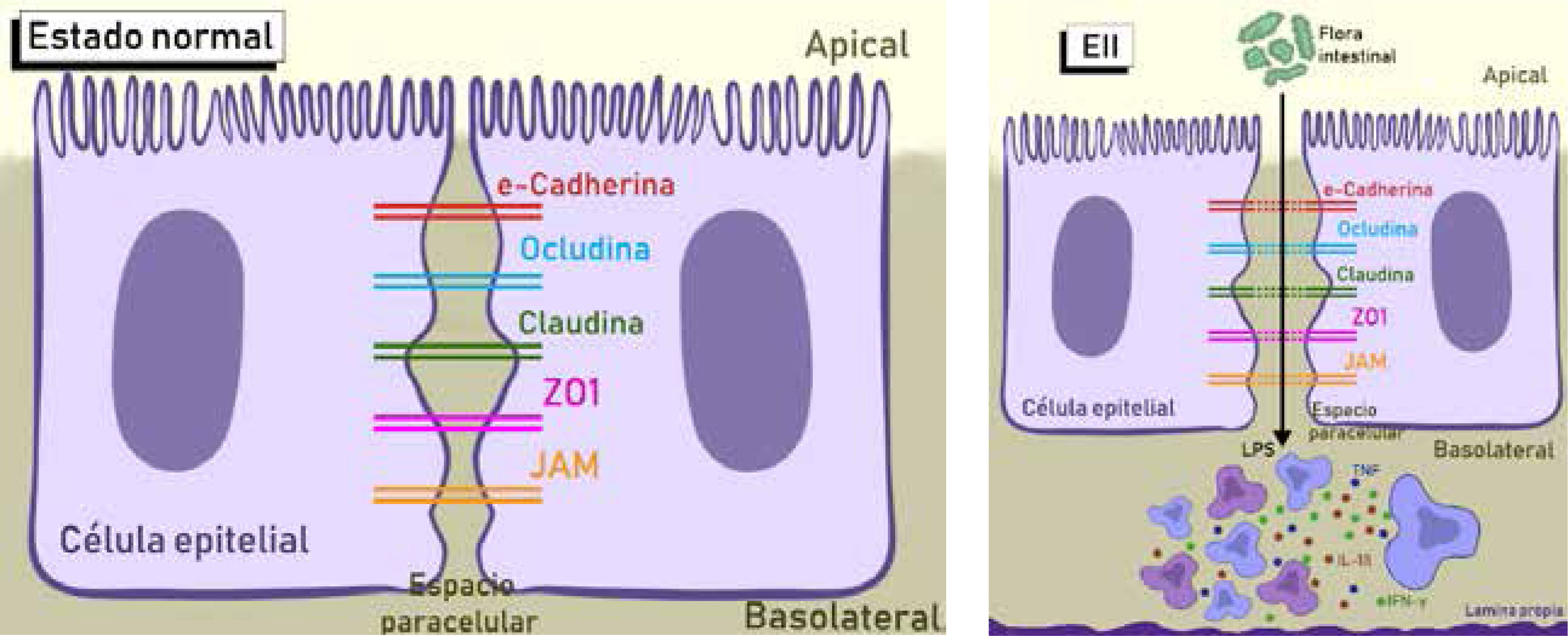
La barrera intestinal incluye el epitelio de superficie, la capa de mucus, los productos secretados como la IgA y defensinas, y la peristalsis efectiva. El epitelio de superficie se compone de una única capa de células epiteliales polarizadas, separadas por un complejo de uniones estrechas que comprenden, entre otras, a la claudina, la ocludina y la tricelulina. Su función es de sellar la membrana y regular el paso de iones y proteínas desde el lumen. Se ha documentado alteración de la expresión de claudinas y ocludinas en modelos de EII, estando su función regulada por la expresión de citoquinas inflamatorias y cambios en la microbiota6. En la figura 4 se muestra un esquema representativo de las alteraciones a este nivel y las consecuencias fisiopatológicas.

2.2. El sistema inmune adaptativo incluye linfocitos T y B, y células dendríticas que confieren respuestas inmunes específicas instruidas por señales derivadas de las células presentadoras de antígeno y los complejos mayores de histocompatibilidad.
Este componente de inmunidad celular del sistema inmune de mucosas se compone de linfocitos T CD8+citotóxicos, células CD4+T helper, que se subdividen en Th1, Th2 y Th17, y células T reguladoras. Existe una activación inadecuada de células T que pueden responder a antígenos luminales7.
Además, se ha observado un aumento de células B activadas y autoanticuerpos, pese a que su relevancia clínica es desconocida. Se sabe que p-ANCA se relaciona con CU y anti-Saccharomyces cerevisiae se relaciona con EC. Estos han sido usados en el pasado en la clínica para diferenciar CU y EC, aunque las pruebas serológicas de estos anticuerpos no tienen sensibilidad ni especificidad, y se han intentado buscar otros. En la figura 5 se observa un esquema del rol de Th17 en EII7.

2.3 Homing leucocitario. Existe una alteración en el llamado homing leucocitario, un complejo proceso mediante el cual los leucocitos son atraídos por el endotelio vascular en el sitio de inflamación, el cual ha sido el objetivo de diversas terapias específicas en EII.
En un estado inflamatorio, los leucocitos entran a la vénula postcapilar y luego se someten a un proceso llamado “rolling”, donde se hacen más lentos y se adhieren a la pared endotelial. Si existe una señal quimiotáctica apropiada, se inicia el proceso de migración transendotelial8.
Las selectinas y sus ligandos son moléculas expresadas en la superficie de los leucocitos y células endoteliales que están sobre-expresadas durante la inflamación y median este rolling. Las proteínas integrinas heterodiméricas se expresan en la superficie de los leucocitos y median la adhesión al endotelio. La especificidad de integrinas depende de sus subunidades; la β1, β2 y β7 se asocian a adhesión leucocitaria. Las integrinas se unen a su ligando en la superficie del endotelio. El heterodímero α4-β7 es el que media el homingleucocitario en el intestino. En la figura 6 se observa este fenómeno.

Existen alteraciones a nivel de citoquinas que son centrales en el desarrollo de la EII, y que protagonizaron el inicio de la terapia biológica en esta enfermedad. El TNF-α es una citoquina pro-inflamatoria que es producida por macrófagos activados, monocitos y linfocitos T. Se demostró que el intestino de pacientes con EC tiene tanto una expresión aumentada de mRNA de TNFα como niveles elevados de su proteína9. Al inhibir el TNF se logró una reversión de un modelo de colitis murino10.
La forma del TNF unido a membrana puede estar aumentado en los macrófagos de la mucosa y se une al receptor de TNF de células T de la lámina propia. Un mecanismo importante de acción de anticuerpos anti-TNF es la inducción de apoptosis de células T, y la respuesta a este tratamiento se correlaciona con el grado de apoptosis lograda. Además, la terapia anti-TNF protege la función de barrera porque disminuye la apoptosis epitelial. En la figura 7 se observa el rol del TNF en la inflamación intestinal.

El ligando relacionado a TNF 1A, TL1A, y su ligando DR3, están aumentados en la EII, y se correlacionan con la actividad de la enfermedad11 y su interacción hace que se incremente la producción de IFN-γ por las células monucleares de la lámina propia, lo que sinergiza con IL-23 para causar la diferenciación de Linfocitos T CD4 hacia células Th17 que producen IL-17 e IFN-γ. Por esto, se ha pensado que esta citoquina juega un rol central en el desarrollo de la EII12.
3.2Vías JAK/STATLas citoquinas pueden activar vías como la JAK/STAT (Kinasa Janus, su transductor de señal y su activador de transcripción) que son centrales a varios procesos tanto innatos como adaptativos e inflamación. Al inhibir esta vía, se pueden “apagar” diversas citoquinas pro-inflamatorias. Por ejemplo, el tofacitinib una nueva molecula prometedora que inhibe funcionalmente al JAK1 y JAK3, se ha aprobado en 2018 para el tratamiento de la CU activa moderada a severa13 (fig. 8):
3.3Receptores de citoquinas.
Una familia de receptores de citoquinas que señalizan a través de esta vía incluyen la IL-12 e IL-3, siendo estas dos citoquinas, heterodímeros que comparten la subunidad p40, que ha sido objeto de terapia biológica con la anti interleuquina como por ejemplo el Ustekinumab14. La IL-12ha mostrado activar inflamación sistémica al inducir células T a diferenciarse en células Th1 productoras de IFN-γ15, mientras que la IL-23 promueve la diferenciación y supervivencia de células Th1716.
Existen alteraciones inmunológicas relacionadas con el reconocimiento de patógenos intracelulares, como las asociadas a polimorfismos de NOD2, comentado en la sección de influencia genética. El NOD2 es un sensor intracelular de patógenos, incluyendo el componente peptidoglicán de bacterias, y se expresa en el compartimiento de la mucosa incluyendo células dendríticas, macrófagos y células epiteliales. Los polimorfismos de NOD2 resultarían en una caída de las vías del aclaramiento de patógenos, incluyendo señales NF-kB y modulación de receptores toll-like (TLR) desde la membrana celular17. El NOD2 también ha mostrado influenciar la secreción de alfa-defensinas antimicrobianas desde las células de Paneth, con niveles reducidos de éstas en muestras de íleon de pacientes con EC. Todo esto favorece una disbiosis y con ello desarrollo de EII.
3.4Autofagia.La vía de la autofagia no había sido considerada previamente en la patogenia de EII. Un GWAS publicado en 200618, mostró que el polimorfismo de ATG16L1 contribuye al desarrollo de la EC. Este gen codifica para una proteína en la vía del autofagosoma que procesa bacterias intracelulares. La autofagia es un mecanismo de defensa bien conservado que consiste en que los lisosomas degradan los organelos y a los microorganismos para efectos de conservación de energía y protección de microbios. Un déficit en el autofagosoma con la consecuente dificultad para la autofagia ha mostrado afectar las defensas contra patógenos de invasión como Salmonella tiphimurium.
3.5InflamasomaFinalmente, otro proceso que ayuda a regular la homeostasis entre la barrera epitelial y la microbiota comensal es el inflamasoma. Este es una red de proteínas que se ensamblan en células mieloides en respuesta a “señales de peligro” en la forma de patrones de reconocimiento de daño. Las proteínas de éste se clivan en formas activas por enzimas proteolíticas, incluyendo la caspasa-1, y resultan en producción de citoquinas inflamatorias, predominantemente IL-1β, IL-18 e IL-3318. El inflamasoma se activa como respuesta a una gran cantidad de estímulos, dentro de los cuales se incluyen muchos patógenos microbianos, enfermedades inflamatorias, neoplasia y desórdenes metabólicos y autoinmunes. La liberación de estas citoquinas desde las células es mediada por la piropoptosis, una forma inflamatoria de muerte celular. Se ha identificado una proteína, la Nlrp3 que está íntimamente ligada a la activación del inflamosoma en modelos de colitis murino19. Este puede convertirse en un interesante objetivo de terapias en la EII en el futuro20.
4Influencias ambientalesLuego de revisar la influencia de la genética, está claro que son necesarias influencias ambientales. Esto se entiende mejor al ver el rápido incremento de EII en el mundo en vías del desarrollo21.
Los GWAS han sugerido que la composición y función de los microbios que existen en el intestino son un factor importante en la patogenia de la EII. Muchas de las vías identificadas se centran en el reconocimiento y respuesta a microbios intestinales en la mantención de la homeostasis, y la pérdida de la tolerancia a la microbiota comensal juega un rol mayor en la patogenia.
Muchos de los otros factores ambientales, como la dieta y el tabaco, son importantes por su impacto en la microbiota intestinal o la respuesta del huésped a la microbiota.
4.1La microbiota intestinalEn estados de salud, el microbioma intestinal se ha compuesto de cuatro familias de bacterias: Bacteroidetes, Firmicutes, y en menor medida Proteobacteria y Actinobacteria22. La concentración de microbios aumenta del estómago hacia el colon donde están la mayor densidad bacteriana, en torno a 1012 células/gramo de contenido luminal. Estas bacterias tienen múltiples funciones que involucran el sistema inmune de mucosas, resistencia a la colonización contra por ejemplo el Clostridium difficile. Estos son necesarios para mantener el sistema inmune de mucosas: un estudio sugirió que ratones libres de microbiota intestinal están más predispuestos a desarrollar una enfermedad inflamatoria intestinal23. Por esto, la microbiota podría ser considerado un órgano en sí mismo.
En EII se han visto cambios dramáticos en la microbiota, los más importantes son una disminución de Bacteroidetes y Firmicutes y un aumento de la Gammaproteobacteria24. La alfa diversidad, o el número total de especies, está disminuida en la EII activa25.
4.2Dieta en la EIIEl progreso en el conocimiento del microbioma y su relación con la EII ha dejado al descubierto que la dieta puede impactar en la composición y funcionalidad de éste. Estudios clínicos pediátricos no controlados han demostrado que varias terapias dietarias como la nutrición enteral exclusiva y dietas de exclusión recientemente desarrolladas, pueden ser potentes herramientas para la inducción de la remisión al inicio de la enfermedad, en pacientes que fallan al tratamiento biológico, como una terapia para las complicaciones de la enfermedad y para reducir los requerimientos de cirugía26.
Patrones dietarios diferentes resultan en diferencias marcadas en las comunidades microbianas intestinales y pueden explicar una diferencia epidemiológica de la EII en el mundo: la dieta africana tiene distinto perfil de microbiota comparado a una dieta baja en fibra y alta en proteína y grasa occidental27. La disbiosis es más prominente en la EC28.
Esta descrito que en pacientes con EII que existe una predominancia de proteobacterias, específicamente cepas individuales de Escherichia coli29. La contribución a la patogenia directa de esta observación es controvertida.
La dieta es uno de los factores más importantes en el microambiente intestinal normal, lo que impacta sobre la composición microbiana y su función, sobre la barrera intestinal y sobre la inmunidad del hospedero. Las alteraciones en grupos de alimentos específicos pueden tener efectos de largo alcance. En estudios murinos, por ejemplo, la deprivación de fibra causará un cambio de especies de microbiota que consume la capa de mucus intraluminal, causando una depleción de ésta y posteriormente una disrupción de la barrera mucosa, activación inmune y daño tisular30. Datos recientes sugieren que los factores más importantes que regulan los cambios de composición de la microbiota son la ingesta calórica, así como de hidratos de carbono y proteínas31. En la figura 9 se esquematiza el posible rol de la dieta sobre la inmunidad y la microbiota, respectivamente, en EII.
5La dieta como intervención terapéutica
Estudios recientes han expandido nuestra comprensión de posibles roles para la terapia dietaria, primariamente en EC. Algunas de estas, como la nutrición enteral exclusiva (NEE), que consiste en el uso exclusivo de fórmulas líquidas sin exposición a otro tipo de alimentación por 6 a 8 semanas, se han usado primariamente para inducir remisión de la EC temprana o como terapia de soporte32.
El efecto no parece solo depender del tipo de fórmula, sino también considera la exclusión de alimentos habituales. Estudios más recientes32 han demostrado que las estrategias nutricionales pueden tener un rol más amplio, manteniendo la remisión, induciendo remisión en pacientes con falla a biológicos y en pacientes con enfermedad complicada. Existe un rol de ácidos grasos de cadena corta que se logran al fermentar ciertas fibras. Estos ácidos grasos tienen un rol evacuador de neutrófilos de los sitios de inflamación, en el que participan resolvinas, lipoxinas y protectinas33.
5.1Vitamina DOtro componente dietario importante en la patogenia de la EII es la vitamina D. En un modelo en ratones knock out para el receptor de vitamina D, éstos tuvieron una mayor alteración de la flora bacteriana, más inflamación colónica y predisposición a desarrollar una colitis, con una disminución de la expresión de e-cadherinas en las uniones estrechas34. Por otra parte, en un estudio poblacional en EE.UU., las personas nacidas en latitudes sureñas del hemisferio norte tuvieron un menor riesgo de desarrollar una EII, y este riesgo se atribuyó en gran parte a las diferencias en niveles de vitamina D35.
3.4 Los ácidos grasos poliinsaturados (PUFA) son otro factor importante en la dieta que influye en la EII. La relación entre omega-6 a omega-3 puede incrementar la susceptibilidad a la EII; en un estudio pediátrico, aquellos niños que consumieron una dieta alta en PUFA omega 6 versus una dieta alta en omega-3, tuvieron un riesgo aumentado de EC36.
5.2Tabaco y EII.El tabaquismo se considera uno de los factores más significativos que alteran el curso de la EII. Pese a que el tabaco es protector en la CU, éste empeora muchos aspectos de la EC37. El humo inhalado modula el microbioma intestinal38. Se ha reportado un incremento de los phyllum Proteobacteria, Bacteroidetes, así como Clostridium, Bacteroides y Prevotella. Por otro lado, los phyllum Actinobacteria y Firmicutes, así como Bifidobacterias y Lactococcos disminuyeron39.
Además, el tabaquismo puede afectar vías de la inmunidad innata. En un estudio, se demostró un aumento en la autofagia de los enterocitos como respuesta a una injuria oxidativa asociada al tabaco40.
6De la fisiopatología a la clínicaComo se ha mencionado anteriormente, hay numerosos ejemplos en EII donde una comprensión más acabada de la fisiopatología de la EII se ha traducido en nuevas opciones terapéuticas incorporadas a la práctica clínica. En la tabla 1 se muestran las estrategias terapéuticas actualmente utilizadas.
Estrategias terapéuticas actuales según blancos determinados en EII
| Blanco | Mecanismo | Terapia |
|---|---|---|
| Reclutamiento celular | Bloqueo de llegada de linfocitos T al sitio inflamado | Anti-integrinas |
| Mediadores inflamatorios | Bloqueo de la señal inflamatoria celular | CorticoesteroidesMesalaminas |
| Interacciones entre Linfocito T y Célula presentadora de antígenos | Activación de células T | AzatioprinaMetotrexato |
| Antígenos | Eliminación del patógenoReintroducción de bacterias beneficiosas | AntibióticosProbióticosTransplante fecal |
| Receptores de membrana y citoquinas | Aumento de citoquinas antiinflamatorias o disminución de citoquinas proinflamatorias | Anti-TNFAnti-IL12/23 |
| Función de barrera y reparación | Aumento de la barrera epitelial y su reparación | ____ |
Existen numerosos ejemplos en EII donde descubrimientos científicos se han traducido en cambios en la práctica clínica.
Adaptado de Hamilton, Matthew J., et al.50.
Las terapias biológicas que incluyen anticuerpos que bloquean TNF, IL-12/23, integrinas y JAK kinasas son los principales ejemplos. Los mecanismos exactos de eficacia de las terapias más tradicionales como el metotrexato, azatioprina, 5-ASA y los corticoides no se entienden completamente pero parecen tener acciones menos selectivas y más amplias sobre la respuesta inmune adaptativa. Se cree que los corticoides bloquean la producción de citoquinas inflamatorias de células T, mientras que la azatioprina bloquea la activación de células T. Con el actual entendimiento de la microbiota y cómo afecta la inflamación intestinal, las terapias futuras posiblemente se dirijan a modificar el microbioma intestinal o los factores en el compartimiento intestinal que responden a ésta. Como no se ha identificado un microorganismo patogénico al cual dirigir una terapia específica, la terapia antibiótica tradicional no ha mostrado ser un tratamiento efectivo para la EII41,42.
En este sentido, una posibilidad es la de repoblar con organismos beneficiosos en forma de probióticos. Hay evidencia de la eficacia del uso de bacterias como el Lactobacillo y Bifidobacterias en la terapia de mantención de CU y Pouchitis43.
El trasplante de microbiota fecal (TMF) está en etapas precoces de evaluación en el tratamiento de EII. Si bien numerosos reportes de casos y estudios de cohorte han descrito el TMF en pacientes con EII durante las últimas dos décadas, el conocimiento en esta materia sigue siendo limitado y se desconoce el rol final de éste. Se requiere de mayor investigación antes que el TMF se considere como una herramienta más en la terapia de esta enfermedad44.
Dado que una barrera epitelial intacta y correctamente funcionando es fundamental para mantener un estado de salud en pacientes con EII, la curación mucosa, evaluada mediante endoscopía, y la remisión histológica, se han convertido en un objetivo clave en los estudios clínicos, las que han mostrado buenos resultados, reduciendo hospitalizaciones, cirugías y otros objetivos clínicos importantes en EII45. Las terapias anti-citoquinas como los anti-TNF pueden ser beneficiosos en parte por sus acciones en la barrera epitelial46.
7SíntesisEs importante reconocer que el estudio de la patogenia de la EII ha avanzado tremendamente en años recientes, coincidiendo con avances en técnicas de experimentación genética y herramientas de análisis informático. Se pueden obtener grandes cantidades de información de perfiles genéticos, composición de microbioma, entre otros. En el futuro se requerirán técnicas para analizar estos datos e integrar mecanismos de enfermedad. Se espera poder obtener el perfil genético de un paciente con EII para predecir la respuesta a ciertas terapias o bien predecir su curso clínico. Es así como por ejemplo, ciertos polimorfismos de NOD2 se han asociado a enfermedad ileal fibroestenótica y necesidad de cirugía temprana en pacientes con EC47. Es posible que se pueda fenotipificar a los pacientes basándose en el mecanismo de enfermedad subyacente, y de esta forma ofrecer una terapia personalizada y a la medida de cada paciente que padece esta enfermedad.
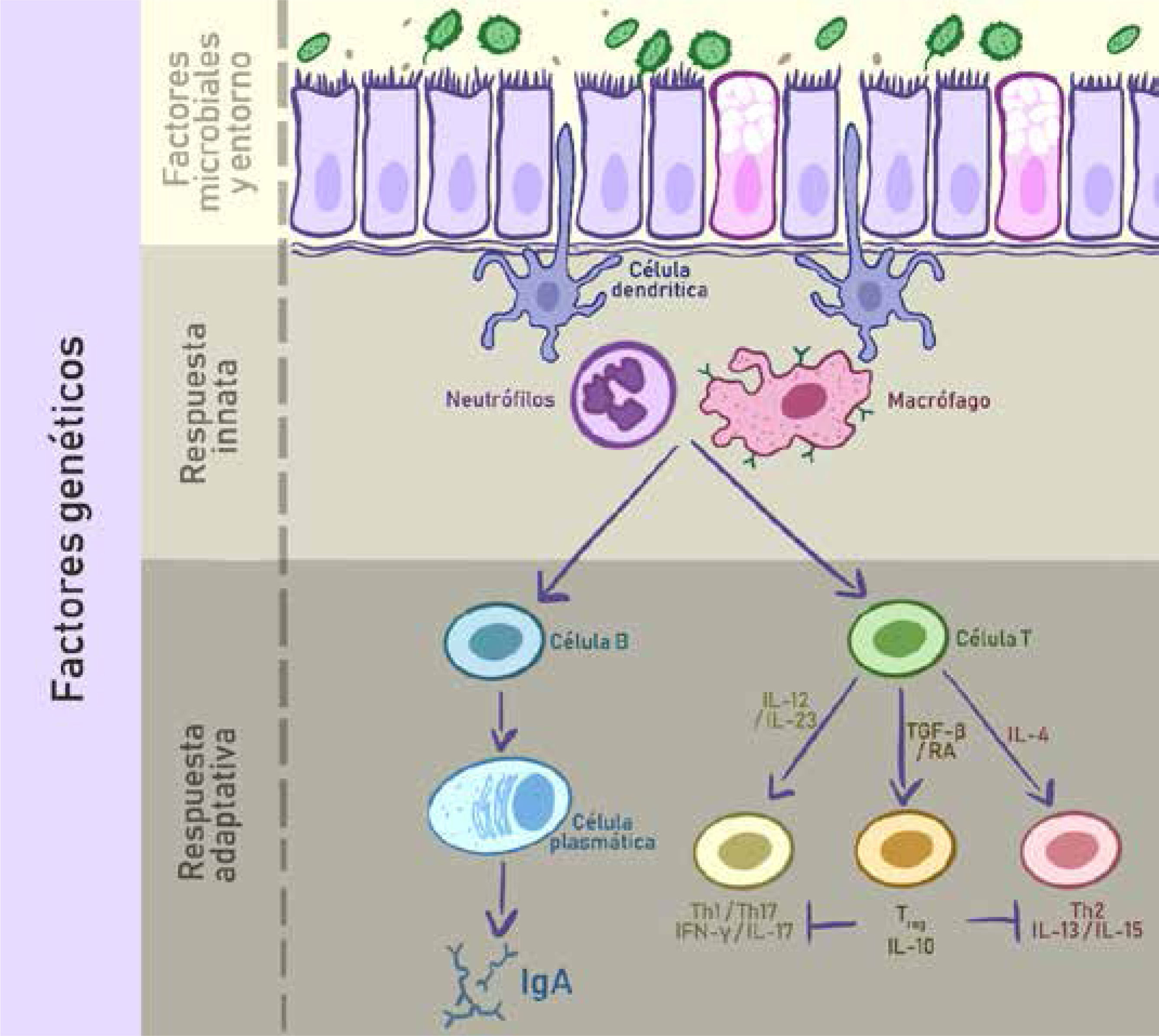
La fisiopatología de la EII es mejor entendida como una relación multidireccional entre factores genéticos, respuestas inmunes -tanto innatas como adaptativas-, factores microbianos y otros ambientales. Las células plasmáticas secretan IgA en la superficie mucosa, y las células T naïve maduran hacia linfocitos T helper tipo 1, 2 o 17 -proinflamatorios-, o bien células T reguladoras -antiinflamatorias-, dependiendo del ambiente local. (IFN-γ: Interferón-γ; IL: Interleuquinas; RA: Ácido retinoico; TGF-β: Factor de crecimiento transformante β; T regs: Linfocitos T reguladores).
Adaptado de Hamilton, Matthew J., et al. “Update on Biologic Pathways in Inflammatory Bowel Disease and Their Therapeutic Relevance.” Journal of Gastroenterology, vol. 47, no. 1, 2012, pp. 1–850.
Estructura del Gen humano NOD2 y sus 3 mutaciones más comunes asociadas a la Enfermedad de Crohn compuesto por CARD (Caspase Activation and Recruitment Domain), NOD (nucleotide oligomerization domain) y LRR (Leucine Rich Repeats).
Adaptado de Yamamoto, Soichiro, et al.2.
En condiciones normales, el epitelio intestinal expresa varias proteínas de uniones estrechas en el espacio paracelular. En la EII, la barrera intestinal se ve comprometida, con una expresión disminuída y una distribución diferente de éstas. La ausencia o disminución de estas moléculas conduce a un aumento en la severidad de la enfermedad al aumentar varias citoquinas pro-inflamatorias, incluyendo IL-1β, TNF-α e IFN-γ secretadas por células inmunes. La microbiota intestinal tiene también un rol importante al regular las uniones estrechas y la integridad de la barrera mucosa. Los patógenos intestinales pueden alterar la función de barrera al incrementar la permeabilidad intestinal a través de mecanismos parecidos.
Adaptado de Castoldi, Angela, et al.48.
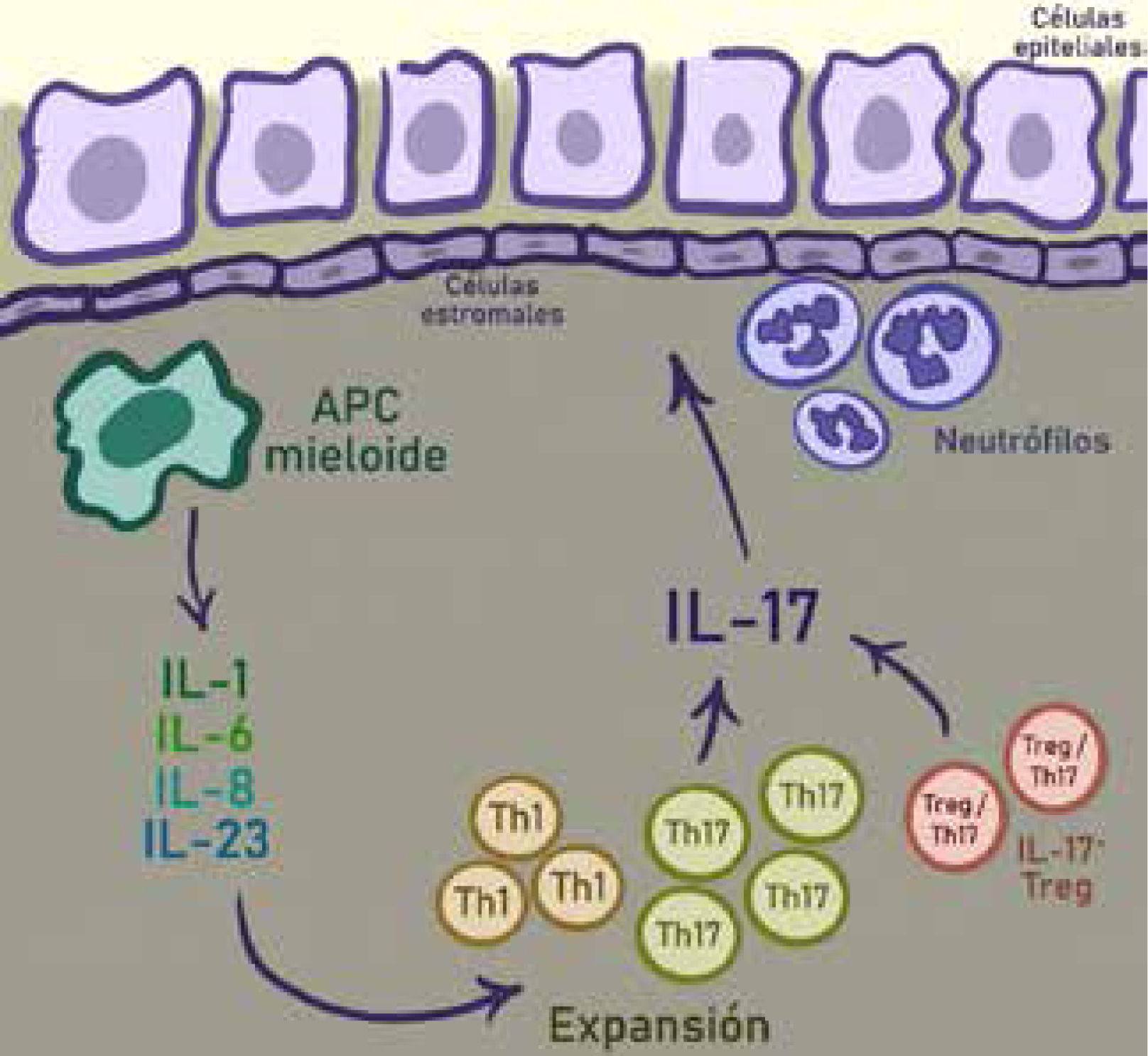
Una gran cantidad de citoquinas inflamatorias se encuentran en el ambiente intestinal, las que son producidas por células presentadoras de antígeno mieloides (CPA), células del estroma, células epiteliales y neutrófilos. Las CPA, junto a IL-1, IL-6 e IL-23 conducen a una expansión de células Th17. Estas células a su vez inducen a las células epiteliales y estromales a expresar más citoquinas proinflamatorias, promover el reclutamiento de neutrófilos y acelerar la inflamación local. Las células Treg/Th17 también se encuentran en el ambiente de la EII, las que suprimen la inmunidad adaptativa de células T pero también secretan citoquinas inflamatorias.
Adaptado de Wilke, Cailin Moira, et al.7.
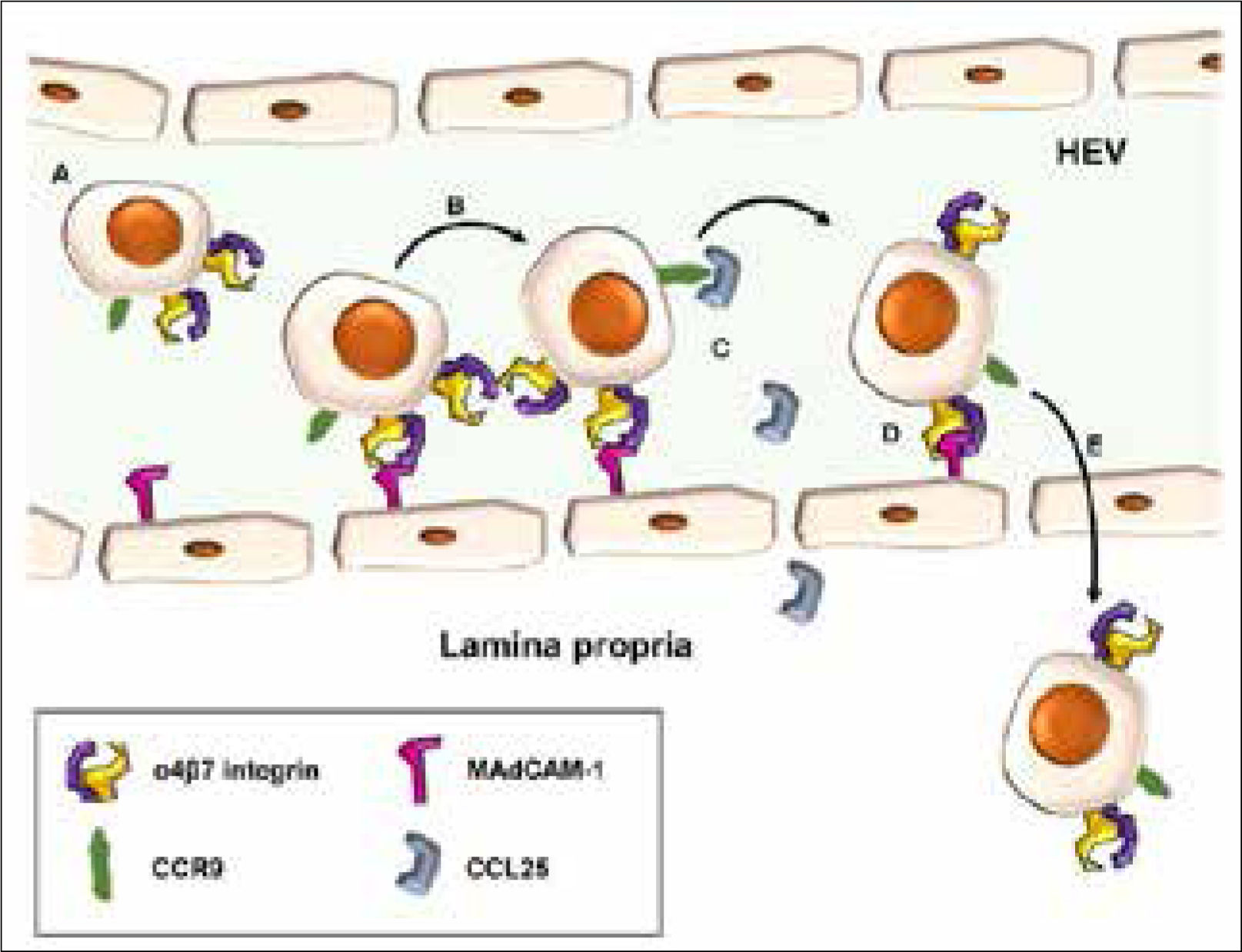
Las células T destinadas al intestino que contienen la integrina α4-β7 y el receptor CC-quimioquina 9 (CCR9) se deslizan por las vénulas de endotelio alto intestinal, a través de interacciones de baja afinidad entre α4-β7 y la molécula de adhesión celular mucosa vascular-1 (MadCAM-1). La activación de CCR9 por CCL25 también es necesaria. Hay una adhesión firme de la célula a la pared endotelial. Se produce una migración transcelular o paracelular hacia la lámina propia.
Adaptado de Zundler, Sebastian, et al.8.
Esta citoquina reguladora ejerce sus efectos a través de 2 receptores: TNFR1 y TNFR2. Los defectos de señalización de TNFR2 son evidentes en una variedad de enfermedades autoinmunes como la EII. Esta señalización activa vías de apoptosis selectiva e inflamación.
Adaptado de Faustman, et al.49.
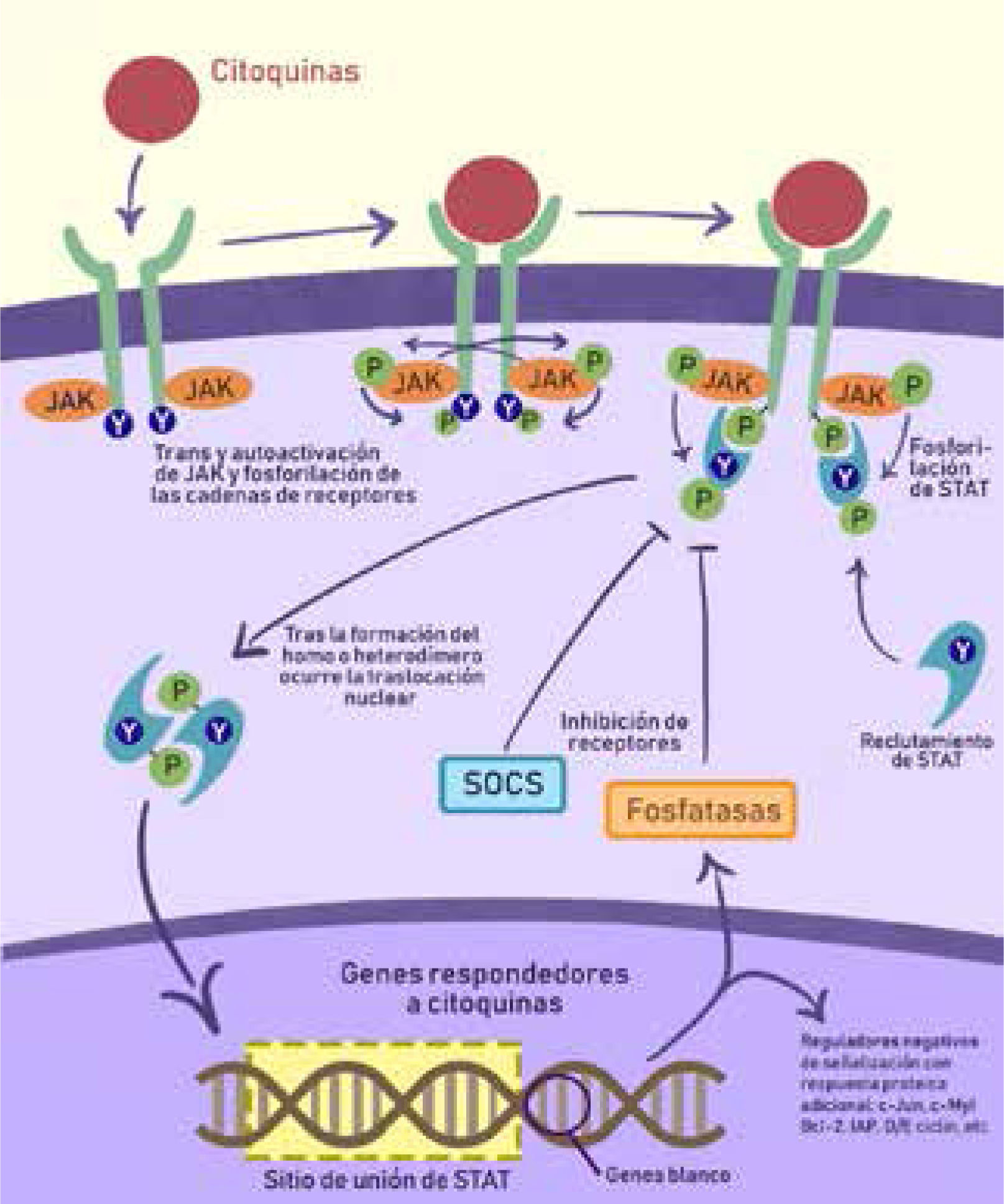
La vía JAK/STAT, activada por citoquinas y su mecanismo de transducción, se ha convertido en un nuevo objetivo terapéutico en EII. Con la unión de un ligando se induce la dimerización de su receptor. Las proteinas JAK ya están pre-asociadas al receptor y luego son auto y transfosforiladas para luego fosforilar las porciones de receptor asociadas. Esto crea sitios de unión para las proteínas STAT. Las STAT son fosforiladas por las proteínas JAK, lo que las libera hacia el citoplasma, donde se aparean como homo o heterodímeros que se translocan al núcleo. Allí funcionan como factores de transcripción al unirse a secuencias específicas de STAT en el DNA. La expresión génica consiste en genes de respuesta general y otros célula-específicos. Existe expresión de proteínas regulatorias y una retroalimentación negativa que previene una señalización excesiva.
(IAP: Inhibidor de apoptosis; JAK: Kinasa Janus; P: grupo fosfato; SOCS: Supresor de señalización de citoquinas, STAT: transductor de señal y activador de transcripción; Y: indicador de residuo de tirosina).
Adaptado de Soendergaard, Christoffer, et al.13.
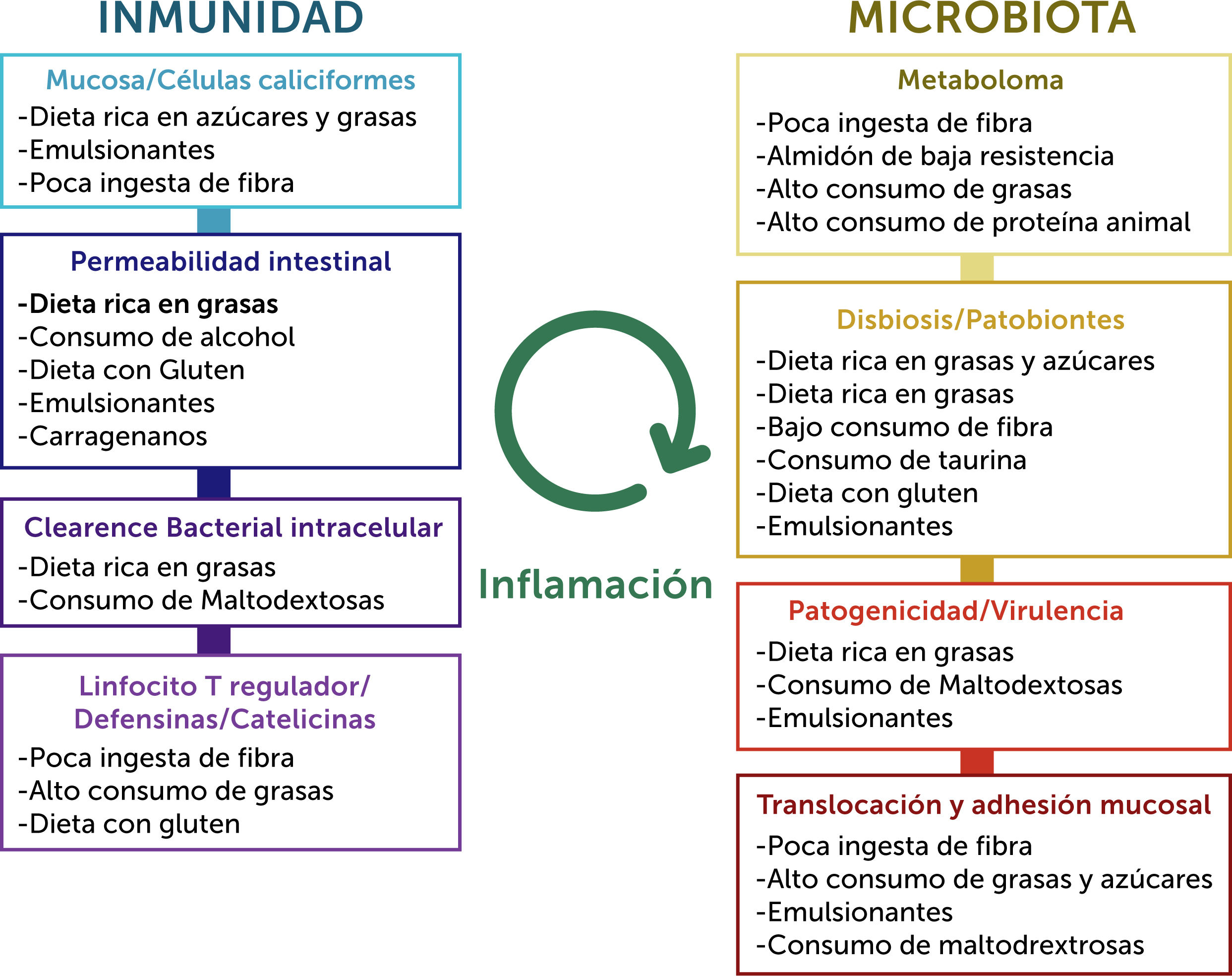
A la izquierda, posibles efectos de factores dietéticos en la barrera del hospedero y en procesos inmunes que conducen a EII. A la derecha, los posibles efectos de estos factores en la microbiota. Ambos procesos convergen centralmente en la generación de inflamación.
Adaptado de Levine, Arie, et al.26.
8Declaración conflicto de interesesLos autores no poseen conflictos de intereses.