




La FPI predomina en el sexo masculino, en edades avanzadas, con tos y/o disnea progresivas. Un 5% se presenta como una forma familiar.
La tomografía axial computarizada de tórax, fundamental en el diagnóstico, en al menos un 50% hace innecesaria la biopsia. El diagnóstico es conjunto con clínicos, radiólogos y patólogos.
La sobrevivencia media es de tres a cinco años desde el diagnóstico. La historia natural es un deterioro progresivo, pero hay formas rápidas y también pueden aparecer exacerbaciones que ensombrecen el pronóstico.
Diversas comorbilidades se han descrito como la hipertensión pulmonar, la asociación con enfisema y el reflujo gastroesofágico. Sólo recientemente aparecen fármacos útiles, que son la Pirfenidona y el Nintedanib. El clásico esquema de prednisona, azatriopina y N-acetil cisteina, se ha demostrado ineficaz. Otros recursos que pueden utilizarse como complementos útiles en la enfermedad son el oxígeno, la rehabilitación, las terapias antirreflujo y el manejo sintomático de la tos.
IPF appears mainly in aged males, with progressive cough and dyspnea. In 5% of the cases the disease presents as a familial form.
CT scan is key in diagnosis of the disease. In no less than 50% biopsy is unnecessary but diagnosis must be made in conjunction with clinician, radiologist and pathologist.
Median survival is 3 to 5 years from diagnosis. Natural history is a progressive deterioration but there are fast evolution cases and exacerbation of the disease that make worse the prognosis.
Pulmonary hypertension, the association with emphysema and gastroesophageal reflux has been described as comorbidities of the disease.
Last year has been published the positive results of therapeuticall trials with two new drugs, Pirfenidone and Nintedanib. The classical regime for IPF with Prednisone, Azathriopine and Acetylcysteine has been showed as useless. Oxygen, Pulmonary rehabilitation, gastroesophageal reflux and cough management are complementary treatment for the disease.
La Fibrosis Pulmonar Idiopática (FPI) es una forma específica de neumonía intersticial fibrosante, progresiva, limitada a los pulmones, que ocurre principalmente en hombres mayores, asociada a hechos radiológicos e histopatológicos, que se expresan en un patrón que puede ser característico llamado de neumonía intersticial usual (UIP).
PRESENTACIÓN CLÍNICAPuede ser bastante característica. Tos persistente, generalmente de meses de evolución, muchas veces instalada después de un cuadro respiratorio agudo en un hombre mayor de 60 años es la queja inicial más común. El examen físico muestra crepitaciones basales bilaterales en más del 70% de los pacientes e hipocratismo digital en alrededor del 20%. El antecedente de tabaquismo se encuentra en la mayoría de los enfermos.
DATOS EPIDEMIOLÓGICOSLa incidencia de la enfermedad no se conoce con certeza. Diversos estudios han propuesto cifras entre 6.8 y 16.3 por 100.000 habitantes. Hay varios estudios de los últimos años, que demuestran un sostenido aumento de la incidencia. La prevalencia tampoco es conocida con certeza, pero un estudio reciente desde los Estados Unidos propone una cifra entre 14 y 42.7 por 100.000 según si se usen criterios diagnósticos estrictos o más amplios. En grupos de edad ≥ de 75 años las cifras de incidencia y prevalencia son de 71 y 271 por 100.000 habitantes para los hombres y 67 y 266 por 100.000 para mujeres.
La mortalidad por FPI ha aumentado en la última década. Usando criterios definitorios rigurosos en los Estados Unidos la mortalidad en 2003 fue de 61.2 muertes por 100.000 habs. en varones y 54.5 en mujeres. En el 60% de los casos la causa de muerte fue progresión de la enfermedad. Otras causas fueron enfermedad coronaria, embolia pulmonar y cáncer pulmonar.
FACTORES DE RIESGOSe han propuesto algunos posibles factores de riesgo, con muy poca evidencia aún al respecto, y con gran dificultad para la interpretación de los estudios correspondientes. El tabaquismo se ha demostrado como tal, con una fuerte asociación con la presencia de la enfermedad. También se ha descrito asociación con la exposición a polvo de metal o de madera, las actividades agrícolas, los peluqueros, cortadores o pulidores de piedras y exposición a ganado y a polvos vegetales o animales. También se han estudiado agentes microbianos especialmente infecciones virales crónicas particularmente virus de Epstein Barr y hepatitis C, pero con resultados contradictorios.
GENÉTICAHay reconocidas formas familiares de la enfermedad. Alrededor del 5% de los casos correspondería a este tipo, cuando más de dos miembros de una misma familia la padecen. El resto son casos esporádicas y no hay diferencias entre ambas formas, salvo que los casos familiares se presentan en edades más tempranas.
Estudios genómicos han sugerido que ELMOD 2, que es un gen de función biológica desconocida, localizado en el cromosoma 4q31, sería un gen de susceptibilidad para la FPI familiar. La transmisión de la enfermedad sería autosómica dominante con penetración variable. La mutación del gen de la proteína C y A2 del surfactante se han asociado a la forma familiar de la enfermedad.
Muy interesante las investigaciones recientes que han documentado variaciones en los componentes del gen de la telomerasa en el 15% de la FPI familiar y en 3% de los casos esporádicos.
PATOGENIALa hipótesis inflamatoria prevaleció por años, pero evidencias de el componente menor de la inflamación en la histopatología de la enfermedad y sobre todo por el fracaso de la terapia antiinflamatoria publicado recientemente, han descartado tal hipótesis que se ha reemplazado por el modelo epitelial fibrótico, siendo las células alveolares epiteliales y los fibroblastos los componentes claves en la patogenia. Es precisamente a este nivel donde se han focalizado todos los intentos terapéuticos de los últimos años.
DIAGNÓSTICOEl diagnóstico de la enfermedad requiere la exclusión de otras causas conocidas de enfermedad intersticial pulmonar que pueden, en alguna etapa de su evolución, dar el patrón radiológico e histopatológico semejante a UIP por FPI (exposición doméstica o ambiental a antígenos orgánicos, enfermedad del tejido conectivo y toxicidad por drogas).
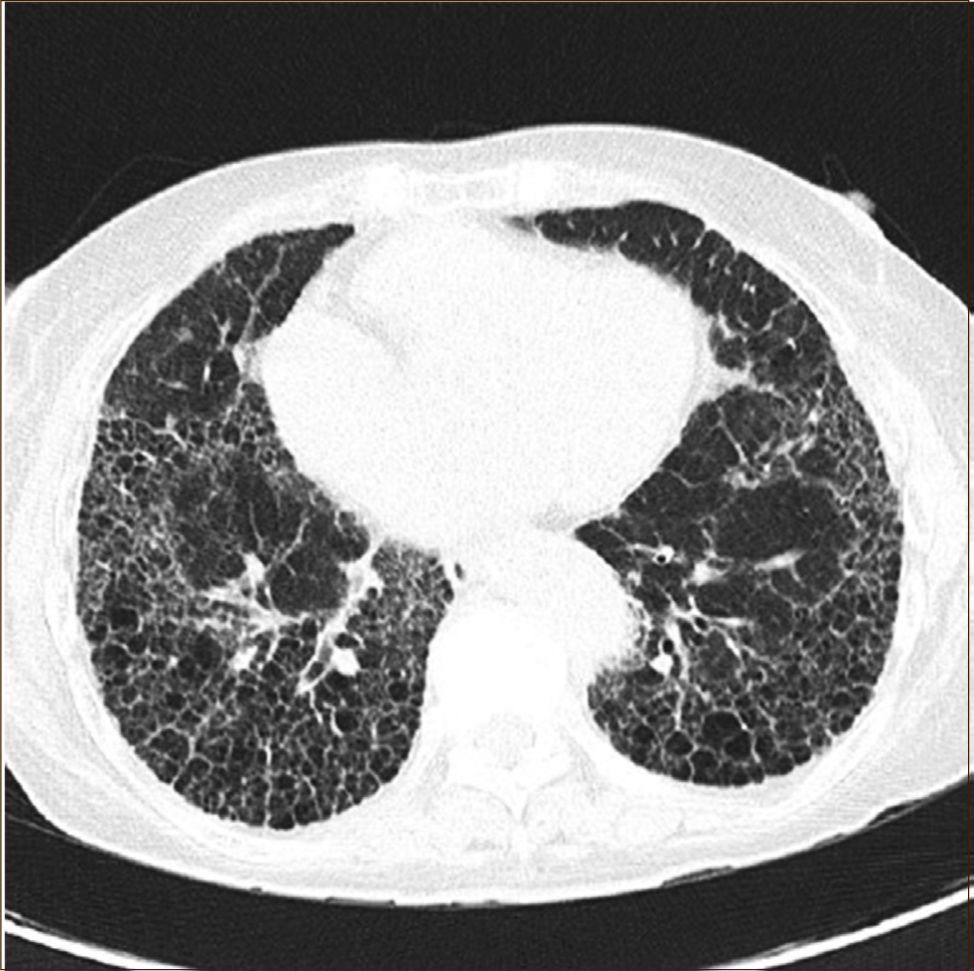
Además exige el patrón radiológico característico de UIP en tomografía axial computarizada de tórax de alta resolución, cuando los pacientes no son sometidos a biopsia quirúrgica pulmonar. Este patrón se expresa con opacidades reticulares a menudo asociadas con bronquiectasias por tracción y con panal de abejas, elemento crítico para hacer el diagnóstico, con distribución basal y periférica y también en parches. El panal de abejas lo constituyen agrupaciones de espacios aéreos quísticos de diámetros comparables de 3-10 mm (ocasionalmente hasta 2.5 cm), subpleurales, con paredes definidas (Figura 1). Puede haber vidrio esmerilado, pero de extensión menor que el reticulado. Adenopatías mediastínicas pueden aparecer, en general menores de 1.5 cm.

Las alteraciones pleurales, los micronódulos, el atrapamiento aéreo, quistes no de panal, extenso vidrio esmerilado, consolidaciones, distribución peribroncovascular son todas alteraciones que sugieren fuertemente un diagnóstico alternativo (Tabla 1).
Criterios de TAC para patrón de UIP
| PATRÓN UIP (requiere los 4) | PATRÓN POSIBLE UIP (sólo 3) | INCONSISTENTE CON UIP (cualquiera) |
|---|---|---|
| Predominio basal y subpleural | Predominio basal y subpleural | Predominio superior o medio |
| Anormalidad reticular | Anormalidad reticular | Predominio peribroncovascular |
| Panal con o sin bronquiectasias por tracción | Extenso vidrio esmerilado > que reticulado | |
| Sin hechos de inconsistencia | Sin hechos de inconsistencia | Micronódulos profusos (bilateral > en lóbulos superiores) |
| Quistes (múltiples, bilaterales, lejos del panal) | ||
| Mosaico difuso de atenuación / atrapamiento en > 3 lóbulos | ||
| Condensaciones segmentarias o lobares |
(Adaptado de referencia 6)
Si el patrón radiológico es de UIP no es necesaria la biopsia pulmonar. Si no lo es, puede estar indicada la biopsia que debe ser quirúrgica, hoy en día a través de videotoracoscopía realizada por un cirujano de tórax experto con muestras de dos sectores diferentes del pulmón, en áreas previamente seleccionadas con ayuda del scanner, lejos de las zonas de panal.
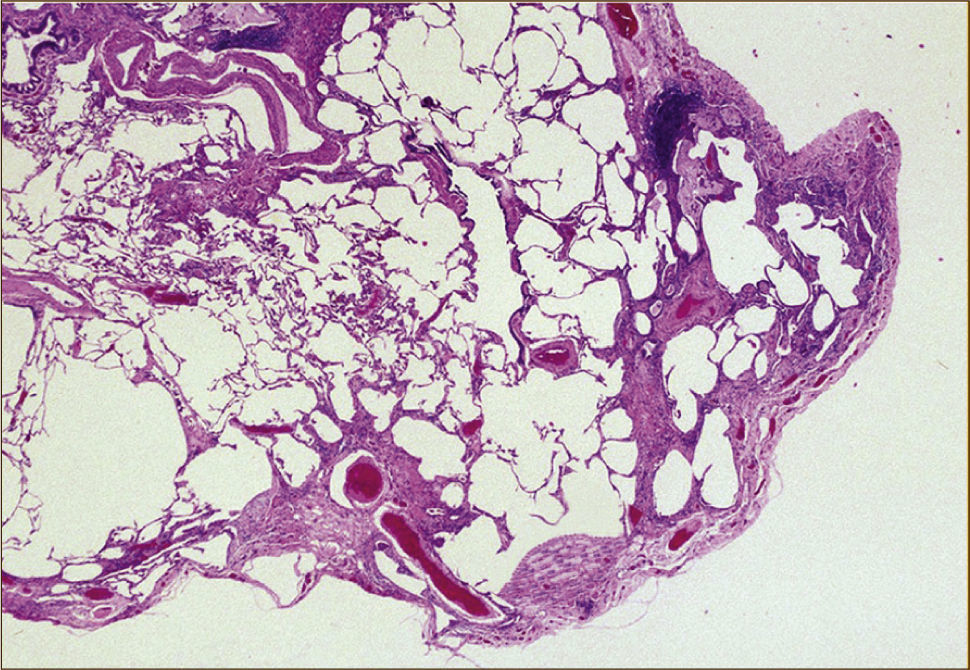
La histopatología de la FPI puede tener un patrón típico que comprende cuatro elementos:
1) Evidencia de fibrosis marcada con distorsión de la arquitectura y panal en una distribución predominantemente subpleural y paraseptal.
2) Presencia de compromiso fibroso en parches.
3) Presencia de focos fibroblásticos.
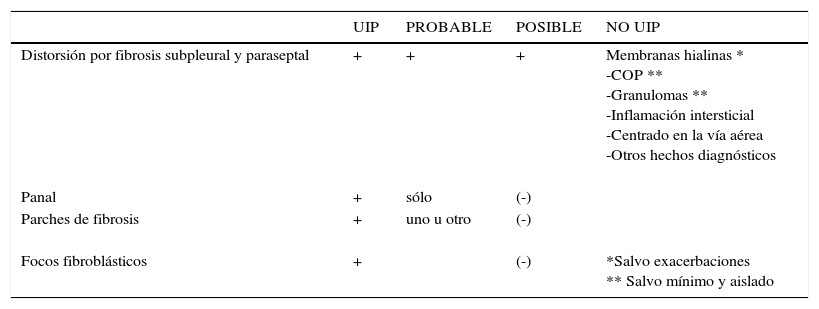
4) Ausencia de hechos excluyentes. Cuando se encuentran estos cuatro elementos el patrón es UIP (Figura 2).

Se habla de una probable UIP si está presente el punto 1, pero sólo el 2 o el 3 y naturalmente el 4. Si no hay 2 y 3 y sólo 1 y 4, se habla de una posible UIP. Si sólo se encuentra panal es una posible UIP. Por el contrario no se trata de una UIP si se encuentran membranas hialinas (salvo en exacerbación), neumonía en organización (salvo mínimo componente), granulomas (salvo uno que otro aislado), infiltración celular inflamatoria prominente lejos de las áreas de panal, cambios centrados en la vía aérea prominentes u otros hechos sugerentes de un diagnóstico alternativo (Tabla 2).
Criterios histopatológicos de patrón de UIP
| UIP | PROBABLE | POSIBLE | NO UIP | |
|---|---|---|---|---|
| Distorsión por fibrosis subpleural y paraseptal | + | + | + | Membranas hialinas * -COP ** -Granulomas ** -Inflamación intersticial -Centrado en la vía aérea -Otros hechos diagnósticos |
| Panal | + | sólo | (-) | |
| Parches de fibrosis | + | uno u otro | (-) | |
| Focos fibroblásticos | + | (-) | *Salvo exacerbaciones ** Salvo mínimo y aislado |
(Adaptado de referencia 6).
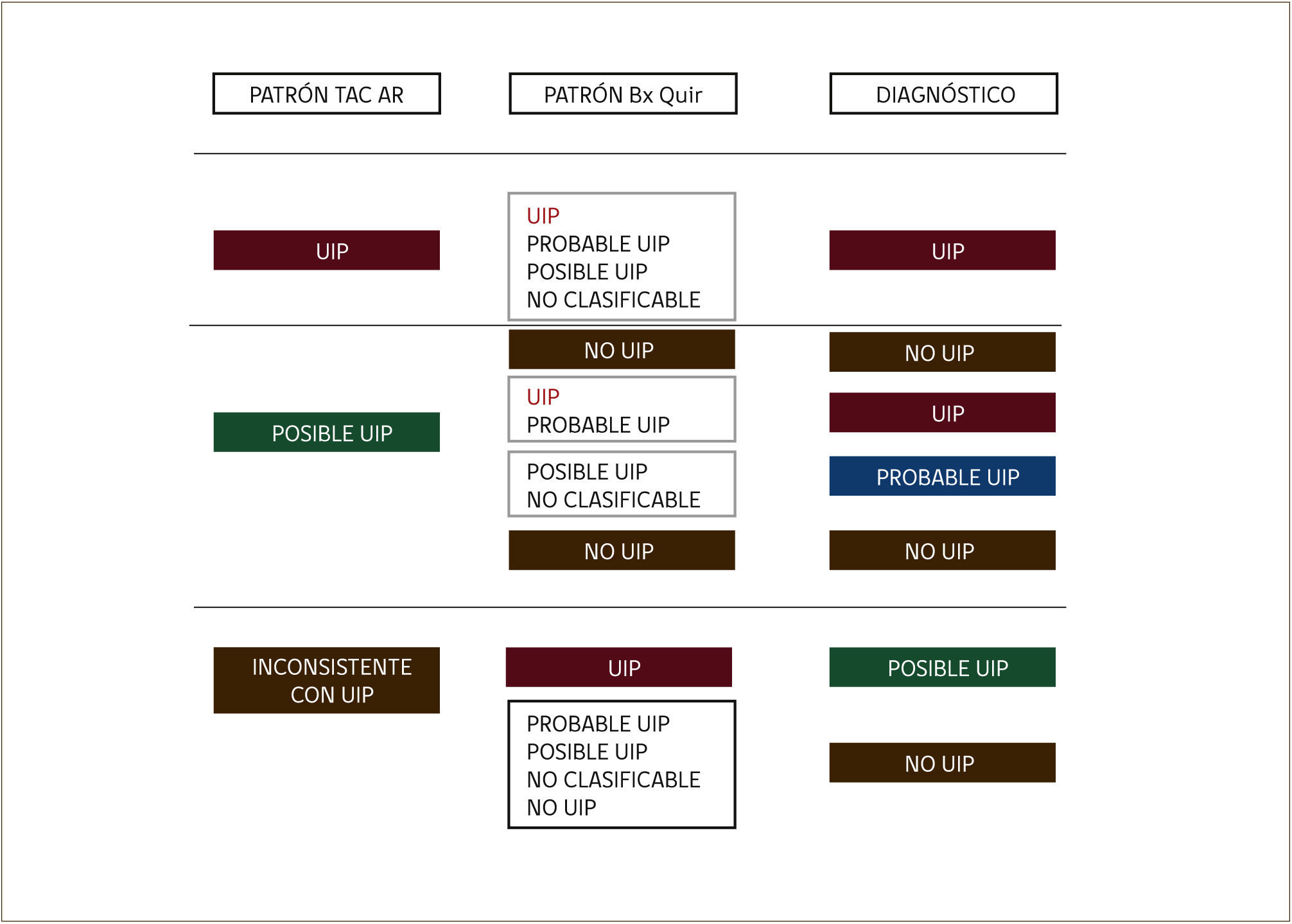
Cuando se dispone de biopsia pulmonar (en alrededor de un 30% de los casos) hay combinaciones específicas entre la imagen radiológica y la biopsia que dan mayor o menor fuerza al diagnóstico (Figura 3).
")
La función pulmonar es la de una alteración restrictiva. La CVF (capacidad vital forzada) está disminuida y la CPT (capacidad pulmonar total) también, con una relación VEF1 /CVF normal o incluso aumentada. La capacidad de difusión del CO está disminuida y es característica la desaturación en ejercicio. En condiciones basales no hay hipoxemia en reposo en las primeras etapas de la enfermedad.
Hay consenso que la precisión de diagnóstico en FPI aumenta cuando se realiza en reunión conjunta con radiólogos, patólogos y clínicos expertos en este tipo de enfermedades. De hecho la radiología y la histopatología pueden ser discordantes; en una misma biopsia pueden haber patologías discordantes y por lo tanto pueden ser necesarios en la decisión diagnóstica antecedentes familiares, de exposiciones diversas, de examen físico y de función pulmonar. Particularmente importante es la posibilidad de una neumonitis por hipersensibilidad crónica, para lo cual pueden ser muy útiles un interrogatorio muy cuidadoso y hechos del lavado broncoalveolar e inducir la necesidad de una biopsia quirúrgica.
De igual modo, se debe investigar la presencia de signos clínicos sutiles de mesenquimopatía (Fenómeno de Raynaud, artritiis, cambios en la piel, motilidad o dilatación esofágica) y la presencia de autoanticuerpos (factor reumatoide, anticuerpo antipéptido citrulinado, título y patrón de anticuerpos antinucleares y más excepcionalmente anticuerpos antisintetasa (Jo-1) creatin quinasa y aldolasa, anticuerpos del Sjögren (SS-A y SS-B) y de escleroderma (scl-70, PM-1)), especialmente en mujeres y en enfermos jóvenes.
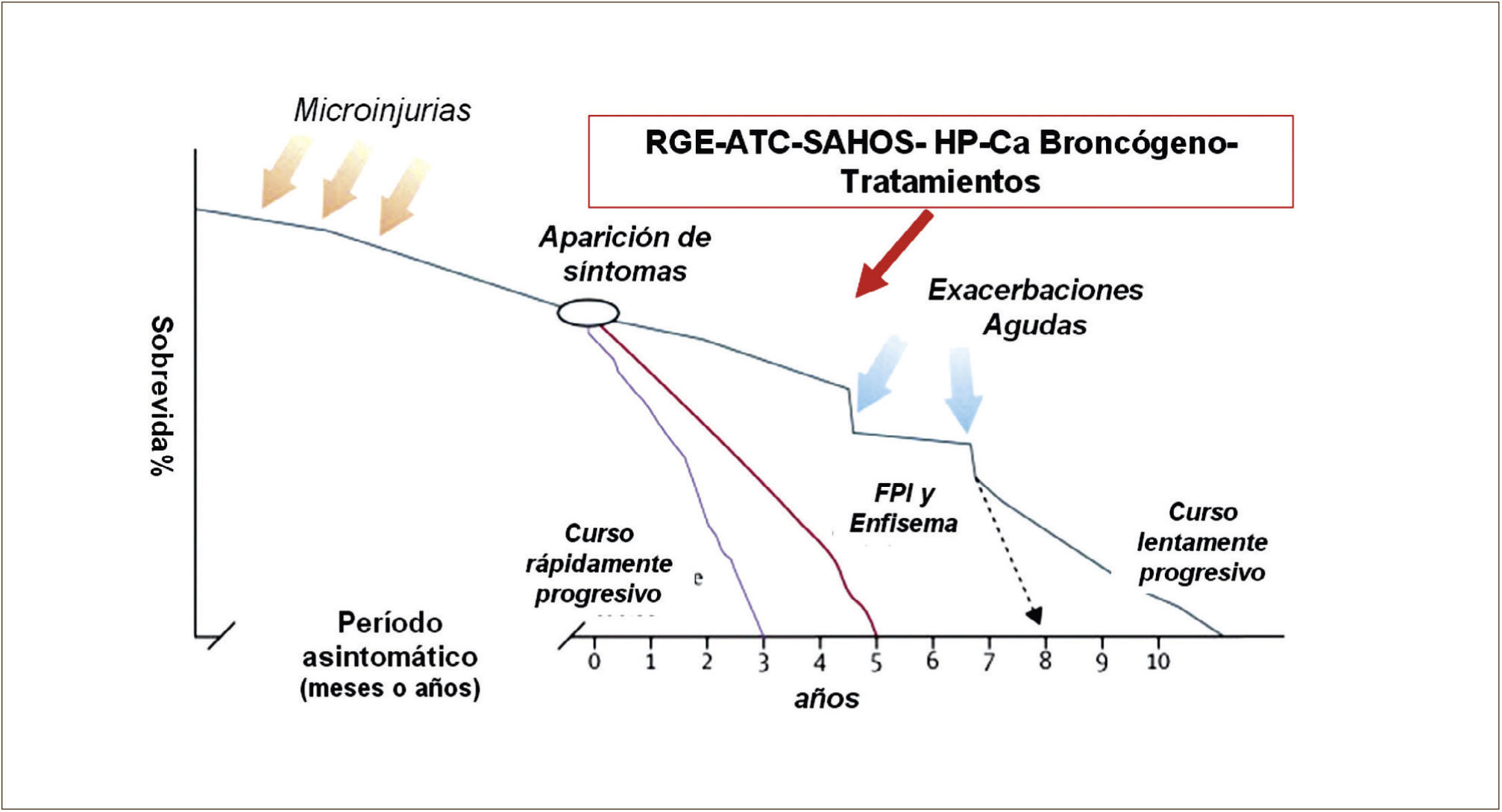
HISTORIA NATURALLa FPI es una enfermedad fatal con una historia clínica variable e impredecible. Diversas publicaciones confirman una sobrevida media desde el diagnóstico de 3 a 5 años. Sin embargo la mayoría de los pacientes evolucionan con una gradual y progresiva pérdida de función respiratoria a través de los años (Figura 4). Una mínima proporción de paciente tiene una evolución extremadamente rápida y existe también un grupo menor que evoluciona muy lentamente. Se ha comunicado diferencias genéticas en el grupo de enfermos de evolución rápida. En algunos pacientes aparecen períodos de agudización o exacerbación, en cualquier etapa de la enfermedad. Esto aparece en el 5 a 10% de los pacientes y se manifiesta clínicamente como un empeoramiento de las condiciones clínicas y funcionales en un período relativamente breve de tiempo (dentro del último mes) sin que eso corresponda a infecciones, descompensación de insuficiencia cardíaca, neumotórax o tromboembolismo pulmonar. En estos casos, al patrón característico de la UIP se agrega nuevas imágenes del scanner que en general son imágenes de vidrio esmerilado difusas o en parches. La histopatología de estos cuadros puede demostrar signos de daño pulmonar agudo, neumonía en organización o un aumento importante de focos fibroblásticos.
PROGRESIÓN DE LA ENFERMEDAD.")
La progresión de la enfermedad se manifiesta por aumento de los síntomas respiratorios, empeoramiento de los índices de función respiratoria, progresión de la fibrosis en el TAC o falla respiratoria y muerte. Se han hecho esfuerzos por definir parámetros que permitan establecer los principales riesgo de mortalidad, lo cual es difícil por la variabilidad que comentábamos en el curso de la enfermedad. La mayor edad y el sexo masculino se han asociado a una menor sobrevida. También el nivel de disnea y sobre todo su empeoramiento en el tiempo se ha asociado a predicción de mortalidad. La función pulmonar se ha estudiado extensamente. El nivel de deterioro de la capacidad de difusión (DLCO), al inicio del estudio, especialmente si éste es ≤ 40% se ha asociado con aumento del riesgo de mortalidad. El deterioro basal de la Capacidad Vital Forzada (CVF), en cambio, no demuestra igual correlación. En cambio la caída longitudinal de CVF en 5 y 10% en 6 meses si tienen clara correlación con el pronóstico. En la DLCO no se demuestra esa correlación en forma consistente. Un cambio en la gradiente alvéolo arterial de oxígeno mayor de 15 mmHg en 12 meses al igual que una disminución de la capacidad pulmonar total en 6 meses también se han mostrado como índices pronósticos. El grado de fibrosis inicial y de panal también tiene un valor pronóstico. Se han propuesto índices compuestos utilizando variables fisiológicas y radiológicas, pero aún faltan estudios prospectivos que los afiancen definitivamente.
En el test de seis minutos se ha sugerido que la desaturación bajo 88% durante la prueba es índice pronóstico. También una capacidad de caminata menor o un tiempo de recuperación de frecuencia cardíaca post test mayores se han asociado con riesgo de mayor mortalidad. Sin embargo, falta aún validación de estos hallazgos.
Los hallazgos histopatológicos se han relacionado con el pronóstico y el número de focos fibroblásticos se ha relacionado con la declinación de la CVF, de la DLCO y con la mortalidad.
COMORBILIDADESPuede haber comorbilidades subclínicas o manifiestas como hipertensión pulmonar, reflujo gastroesofágico, apnea de sueño, obesidad y enfisema.
La hipertensión pulmonar (HP), entendiendo como tal la presión media > 25 mmHg (más de 17 según otros) se ha asociado con aumento del riesgo de mortalidad, al igual que el aumento de la resistencia vascular pulmonar. Se presenta con relativa frecuencia en esta enfermedad y hay relación con una baja DLCO, con una distancia recorrida más corta en el test de seis minutos y con desaturación durante el ejercicio. Sin embargo tiene poca correlación con los índices de restricción pulmonar.
Desde el año 2005 Cottin describió la asociación de fibrosis pulmonar y enfisema (CPFE) como un síndrome resultante de la asociación de distintos hechos, incluyendo el hábito tabáquico, severa disnea, hallazgos espirométricos relativamente leves, capacidad de difusión muy reducida, hipoxemia al ejercicio y hechos radiológicos característicos, además de una alta probabilidad de hipertensión pulmonar. Estos pacientes tiene un pronóstico mucho peor que los pacientes con FPI sin enfisema.
La asociación de hernia hiatal y reflujo gastro esofágico con FPI ha sido bien documentada. Se sabe que esta patología digestiva es más frecuente en edades avanzadas y sus síntomas clínicos son menores. También la FPI es una enfermedad de la edad avanzada y por lo tanto su asociación es explicable. Se ha postulado la microaspiración como un factor etiológico en la patogénesis de la FPI, pero no se han demostrado hechos histopatológicos de aspiración en áreas de UIP lo cual ha puesto dudas al respecto. Sin embargo hay diversas comunicaciones clínicas que sugieren una unión entre la supresión del RGE y la estabilización de la enfermedad fibrosante. Se ha comunicado la detección de biomarcadores de aspiración, tales como la pepsina, en las secreciones bronquiales y especialmente en LBA en pacientes con FPI con exacerbación, planteándose la hipótesis que estarían relacionados con su patogenia.
MARCADORES DE ENFERMEDADHay aún pocos datos sobre marcadores de LBA y séricos en FPI. KL-6 (Krebs Von den Lungen) es una glicoproteina de alto peso molecular clasificada como una mucina humana MUC1 que es producida por neumocitos tipo II en regeneración. Sus niveles séricos están aumentados en la FPI y podrían correlacionarse con el riesgo de mortalidad. Semejantes datos hay de proteína A y D, quemoquina CCL-18, péptido natriurético cerebral. En plasma y en LBA de las FPI las metalproteinasa de matriz MMP1 y MMP7 están aumentadas y se correlacionan con la severidad de la enfermedad. Los niveles del SP-A en LBA parecen correlacionarse con la sobrevida y la presencia de fibrocitos circulantes están asociados con peor sobrevida.
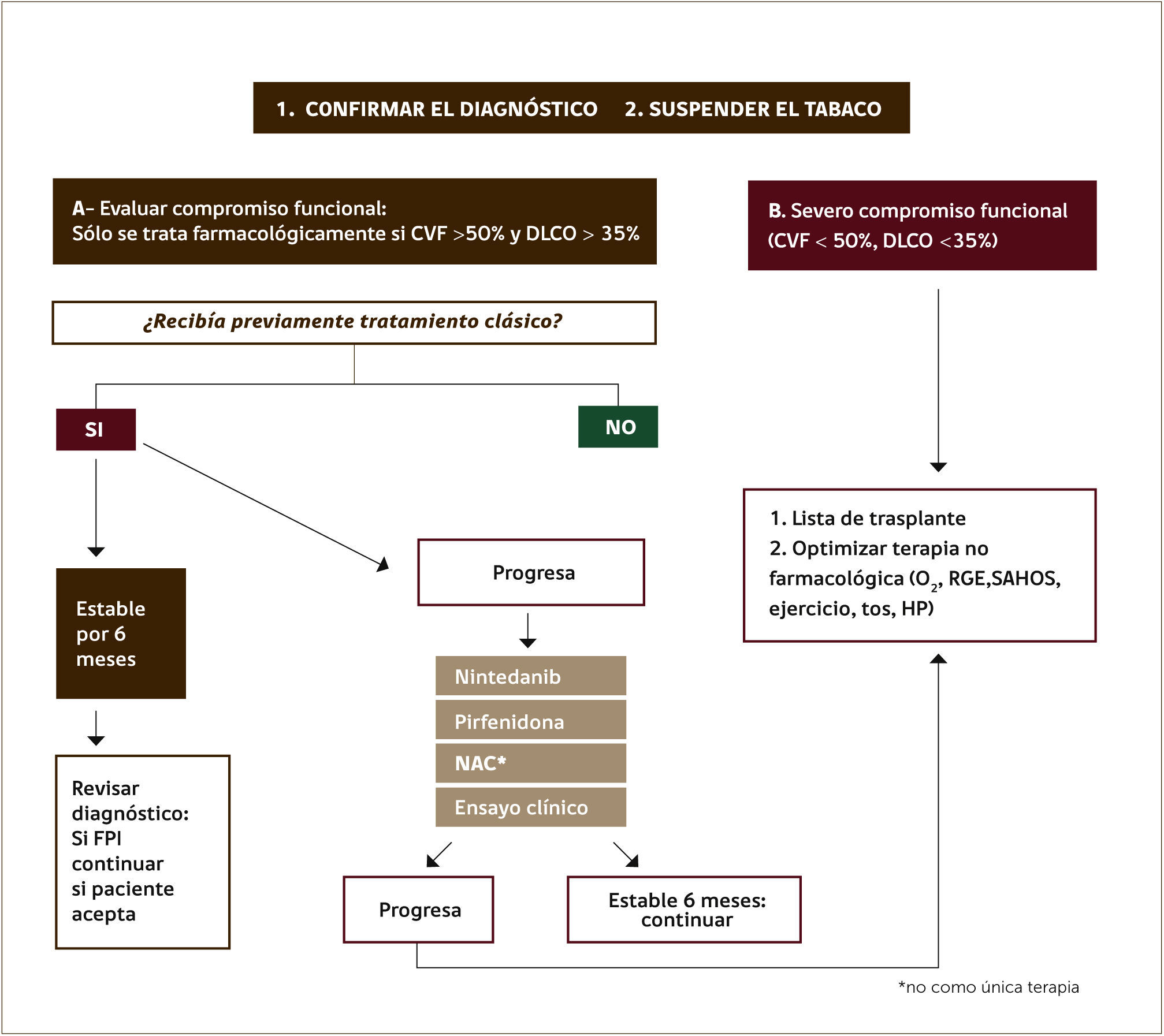
TRATAMIENTOLa terapia actual de la FPI intenta detener el proceso de fibrosis progresiva, aliviar los síntomas que produce la enfermedad e identificar y tratar las complicaciones.
Varias investigaciones recientes sugieren fuertemente que la FPI son varias enfermedades y por lo tanto es desafío del futuro identificarlas y posiblemente lograr diferentes esquemas y algoritmos para tratar sus diferentes tipos. Por esto las terapias del futuro seguramente abarcarán varios blancos.
El año 1999 se publicó el primer consenso internacional de diagnóstico y tratamiento de esta enfermedad y sólo el año 2004 se realizó el primer estudio randomizado de terapia de FPI.
El año 2011 las Sociedades de Enfermedades Respiratorias de Europa, AUA, Asia y Latinoamericanas, publicaron las recomendaciones terapéuticas basadas en la evidencia para diagnóstico y tratamiento de la FPI. En dicha reunión se recomendó no tratar pacientes con FPI con monoterapia corticoesteroidal, práctica que aún vemos en nuestro medio. Tampoco se recomendó el tratamiento con colchicina, medicamento que se usó universalmente durante un tiempo. Sobre la clásica combinación de terapia con prednisona, azatriopina y N acetil cisteina, que se ha usado universalmente por más de 20 años, la recomendación fue menos categórica. En general se recomendó no usar esta combinación, sin embargo, admitió existir un grupo de pacientes para los cuales sería una alternativa razonable. Desde esa fecha hasta el momento el mayor conocimiento de la fisiopatología y las experiencias terapéuticas han estimulado la aparición de diversas guías nacionales que han diferido parcialmente de las recomendaciones del consenso del 2011.
En la FPI, como en todas las enfermedades crónicas, existe un tratamiento farmacológico específico, que en esta enfermedad es por ahora muy limitado, y un tratamiento general que apunta al control de síntomas y comorbilidades. Esta segunda parte de la terapia es muchas veces lo que podemos ofrecer a nuestros pacientes y por tal debe dársele especial importancia.
TRATAMIENTO FARMACOLÓGICOEn mayo de 2012, Raghu y cols publicaron un estudio patrocinado por el Instituto Nacional de Salud de EE.UU., sobre “Prednisona, Azatioprina para la fibrosis pulmonar”, el llamado Estudio Panther. Este fue el primer estudio que comparó la triple terapia clásica de Prednisona, Azatriopina y N-Acetil cisteina (NAC) contra placebo, con tres ramas: placebo, la triple terapia y sólo NAC. El estudio demostró mayor mortalidad e ingresos hospitalarios en el grupo de pacientes que recibieron la triple terapia versus los que recibieron placebo. La rama del estudio que compara la NAC contra placebo, se publicó independientemente en mayo de 2014 concluyendo que la NAC no ofrecía beneficios sobre el placebo, con respecto a la preservación de la capacidad vital forzada (CVF) en pacientes con FPI con leve a moderado compromiso de la función pulmonar.
Estos estudios han producido gran revuelo entre los que se dedican a esta enfermedad y aunque se han publicado comentarios que discuten los resultados aparentemente tan categóricos del Panther, la mayor parte de los consensos nacionales recientes están de acuerdo en no iniciar este esquema en pacientes que recién se diagnostican. Una situación diferente son los pacientes que han estado largo tiempo con este esquema, con buena tolerancia y aparente estabilización de la enfermedad. A pesar que no podemos certificar que dicha estabilización sea debida a la terapia y no al curso natural de la enfermedad en determinado paciente, es razonable, y con un conocimiento acabado y aprobado por el paciente, seguir con el mismo esquema.
Durante los últimos años se han ensayado múltiples fármacos con diferentes racionalidades terapéuticas en FPI, en ensayos bien realizados, controlados en series grandes de pacientes, en grupos randomizados con técnicas de doble ciego placebo control. Así ha ocurrido con el Interferon γ 1β, Bosentan, el Ambrisentan y el Macitentan, antagonistas de la endotelina A y B, el Etanercept, un anti TNF, los anticoagulantes, el Everolimus, derivado de la Rapamicina, el Sildenafil, el Imatinib que es un inhibidor de tirosin quinasa con actividad contra los receptores de los factores de crecimiento derivado de las plaquetas, con resultados generalmente negativos.
Hay varias moléculas bajo estudio actualmente, como por ejemplo la Anti IL-13 pues IL-13 es un potente estimulador de la proliferación fibroblástica y de la síntesis de matriz extracelular, una anti CCL2 que es una proteína quemotáctica de los monocitos, basófilos, células T, células dendríticas inmaduras y fibrositos. Por último hay interesantes ensayos en curso de moléculas anti TGFβ. El TGFβ es una citoquina profibrótica presente en todas las células pulmonares, con múltiples funciones muy relacionadas con el fenómeno fibrótico.
Sin embargo, hay dos moléculas cuyos ensayos terapéuticos han resultado positivos y que también recientemente se han publicado sus alentadores resultados. La primera es la Pirfenidona, que es una piridona con efectos antiinflamatorios, y antioxidantes con antagonismo de los efectos del TGF β1 “in vitro”. Actúa también como un antifibrótico alterando la expresión, síntesis y acumulación de colágeno, inhibiendo el reclutamiento, proliferación y expresión de las células productoras de matriz extracelular. Los primeros estudios sugirieron beneficio de pacientes IPF tratados con el medicamento. Posteriormente hubo dos comunicaciones desde Japón; la primera se suspendió por el comité revisor, por encontrar significativamente menos exacerbaciones de la FPI en el grupo tratado. La segunda comunicó el hallazgo de una menor caída de capacidad vital forzada (CVF) en el grupo tratado y también una diferencia positiva en la sobrevida libre de enfermedad, ambas con significación estadística. Hubo sin embargo objeciones metodológicas a este ensayo. Hubo un tercer estudio internacional que comprendió dos ensayos semejantes paralelos: uno de ellos cumplió su objetivo primario con un resultado positivo significativo de menor caída de CVF en el grupo tratado. El otro ensayo sin embargo no cumplió este objetivo. Un último estudio fue publicado en mayo 2014 que randomizó 555 pacientes con la enfermedad a recibir Pirfenidona oral 2403mg diarios por 52 semanas o placebo y los resultados fueron francamente positivos. Hubo una reducción del 47% en la proporción de pacientes que tuvieron una declinación absoluta de su CVF, del 10% o más en el porcentaje de la CVF ideal o que fallecieron y también hubo un relativo aumento del 132.5% en la proporción de pacientes sin declinación de CVF, una reducción en la caída de la distancia caminada en seis minutos y una mejoría en la sobrevida libre de progresión de enfermedad. Los efectos laterales de la droga fueron fundamentalmente digestivos (náuseas y vómitos, dispepsia, baja de peso), pero fueron calificados como leves o moderados y llevaron a discontinuar la droga en sólo el 2.9% de los tratados. Hubo también rash cutáneo en el 28% vs 9% en el grupo placebo pero se conoce la fotosensibilidad que puede provocar la droga.
La segunda molécula cuyos estudios terapéuticos han resultado positivos y que también han sido publicados recientemente es Nintedanib. Es una molécula del grupo de las tirosin kinasas que inhibe tres kinasas: del receptor de factor de crecimiento derivado de las plaquetas, del factor de crecimiento vascular endotelial y del factor de crecimiento de los fibroblastos. La inhibición de estos tres factores podría reducir el proceso fibrótico. Se publicó un estudio fase II en el cual se comparó tres dosificaciones y placebo en 432 pacientes. La dosis mayor redujo la declinación de la CVF, el número de exacerbaciones y mejoró la calidad de vida. Esto fue asociado a moderados efectos gastrointestinales y hepatotoxicidad. Estos resultados positivos fueron muy estimulantes y dieron pie a un nuevo estudio internacional fase III, en dos grupos grandes de pacientes cuyos resultados acaban de publicarse, nuevamente en la prestigiosa publicación “The New England Journal of Medicine” en mayo de 2014. Fueron dos estudios paralelos el IMPULSIS 1 y el IMPULSIS 2 que randomizaron 1066 pacientes. La caída anual de la CVF fue de 114.7 ml en el grupo tratado vs 239.9 en el grupo placebo en IMPULSIS 1 y 113.6 vs 207.3 en IMPULSIS 2 y las diferencias fueron significativas. Hubo sí resultados discordantes entre ambos estudios en relación al tiempo a la primera exacerbación que no fue diferente en el primero y si fue ventajosa para la droga en el segundo. En relación a los efectos laterales, la diarrea fue un problema universal en ambos estudios con diferencias importantes con el grupo placebo (61.5 vs 18.6 y 63.2 vs 18.3). Sin embargo este síntoma sólo llevó a discontinuar la droga en menos del 5% de los pacientes.
Estas dos moléculas han sido recientemente aprobadas por la FDA en EUA. El Nintedanib aún no está disponible para su venta pero próximamente lo estará. En la mayoría de los países latinoamericanos aún no se cuenta con la Pirfenidona y debe importarse a un costo alto, pero es posible conseguirla. Una reciente recomendación para el diagnóstico y tratamiento de la FPI de la Asociación Latinoamericana de Sociedades de Tórax (ALAT) recomienda el siguiente algoritmo de terapia. (Figura 5).
TRATAMIENTO NO FARMACOLÓGICO.")
Supresión del tabaco: La FPI es más frecuente en fumadores y se han propuesto diversos mecanismos por los cuales el cigarrillo podría participar en las repetidas microinjurias del epitelio respiratorio. Es por lo tanto indispensable que el paciente deje el tabaquismo al momento del diagnóstico.
Oxígeno: Es característico del trastorno funcional de estos pacientes que presenten hipoxemia de ejercicio, trastorno que se va profundizando a medida que la enfermedad progresa. En etapas relativamente avanzadas de la enfermedad los pacientes pueden presentar una saturación basal de la hemoglobina muy poco alterada, pero cae en forma manifiesta con poco ejercicio y esta caída debe ser el parámetro usado para la indicación de oxigenoterapia. Hay pocos datos en la literatura para avalar esta indicación y por lo menos un estudio retrospectivo no encuentra ventajas en la sobrevida. No obstante es experiencia compartida por muchos médicos que tratan estos pacientes la mejoría de capacidad de ejercicio y de su calidad de vida al utilizarlo. El consenso que estamos aludiendo de 2010-2011, lo recomendó con una fuerte indicación pero sin unanimidad.
Rehabilitación: En general se han estudiado programas de rehabilitación complejos que incluyen condiciones aeróbicas, ejercicios de fuerza y flexibilidad, charlas educativas, intervenciones nutricionales y soporte psicosocial. Estos programas han demostrado mejorías en la capacidad de caminata y en la calidad de vida y el consenso los recomienda con una indicación débil. Sin embargo, estos programas son caros, requieren el concurso de kinesiólogos y no están disponibles en muchas partes. A pesar de ello, aunque no estén disponibles, el médico tratante tiene el recurso de estimular e insistir al paciente que, a la medida de sus posibilidades, mantenga un plan de ejercicios simples y que camine con oxígeno, lo cual puede paliar las condiciones de atrofia muscular que muchos de estos pacientes tienen en etapas más avanzadas de su enfermedad.
Trasplante pulmonar: La experiencia mundial y de varios centros en Latinoamérica demuestra que la sobrevida del trasplante pulmonar en pacientes con FPI es 50-60% a los cinco años. Estas cifras superan largamente las expectativas medias de sobrevida de los pacientes fibróticos y por lo tanto avalan el valor de esta indicación. El trasplante pulmonar sin embargo es una terapia cara, que sólo se realiza en pocos centros y que requiere que el candidato permanezca cerca del lugar donde se realiza el procedimiento, antes y después del trasplante por plazos no menores a un año. Por lo tanto, debe considerarse muy en forma muy prudente esta indicación para evitar viajes, gastos y falsas expectativas en los pacientes.
TRATAMIENTO DE COMPLICACIONES Y COMORBILIDADES FRECUENTESVentilación mecánica: En el consenso de 2010, se reportó varios estudios con número limitado de paciente con altísimas mortalidades en estas condiciones. La recomendación general fue no usar este tipo de ventilación, aunque admitió como una elección razonable en una minoría de pacientes. La ventilación no invasiva puede ser una opción y en casos excepcionales la invasiva puede ser puente para el trasplante.
Exacerbaciones agudas: La terapia usualmente recomendada son altas dosis de corticoides, específicamente 1gr de metilprednisolona diaria por tres días consecutivos para posteriormente seguir con altas dosis de corticoides orales, con progresiva reducción de las dosis. La recomendación es débil, sin evidencias más que anecdóticas y justificada por la alta mortalidad del cuadro.
Hipertensión pulmonar (PA media > 25 mm Hg): Epoprostenol y Bosentan se han utilizado en pequeñas series de pacientes. Sildenafil, en grupo pequeño de pacientes ha demostrado mejorar el test de caminata y la hemodinamia pulmonar. El Consenso de 2010 sugiere un ensayo de terapia vasomoduladora en hipertensiones severas pero, como una débil recomendación dado la pobre calidad de la evidencia disponible.
Reflujo gastroesofágico: Es muy prevalente en FPI y en la mitad de los casos asintomático. La recomendación del consenso internacional es muy vaga. Recomienda con un débil énfasis tratar en la mayoría de los pacientes asintomáticos sin especificar de qué mayoría se trata. Hay fundamentos muy pobres en la literatura y hay riesgo potencial de neumonías y de osteoporosis con el tratamiento crónico del RGE. Sólo la mitad de los votantes en la reunión del Consenso de 2010 fue proclive de tratar. Es un terreno que hay mucha discusión. Muy recientemente se publicó un polémico trabajo que afirmaba, luego de una investigación retrospectiva de bases de datos sobre pacientes con terapia anti RGE y FPI que la terapia anti RGE está asociada con sobrevida más larga en FPI. Este trabajo ha sido criticado. Sí hay acuerdo en que claramente se requiere mayor investigación del problema. El autor declara no tener conflictos de interés, en relación a este artículo.