






Las enfermedades pulmonares intersticiales (EPI), son un grupo heterogéneo de trastornos, caracterizados por inflamación y/o fibrosis, que afectan el intersticio pulmonar. Históricamente, se basaban en características histopatológicas de biopsias pulmonares, pero con el avance de la tomografía computarizada de alta resolución (TCAR), el enfoque ha cambiado, permitiendo identificar patrones de enfermedad que sugieren distintas etiologías. La clasificación de las EPI ha evolucionado, y actualmente se clasifican según etiología, patrones histopatológicos y/o radiológicos y curso evolutivo. Las neumonías intersticiales idiopáticas (NII) son un grupo de EPI de origen desconocido, y la fibrosis pulmonar idiopática (FPI) es la más común dentro de las NII. Se ha desarrollado el concepto de enfermedad pulmonar intersticial fibrosante progresiva (EPI-FP), que describe un fenotipo de EPI con progresión a pesar del tratamiento. El diagnóstico se basa en una historia clínica detallada, examen físico, pruebas de función pulmonar, serología autoinmune y técnicas de imagen como la TCAR. El tratamiento varía según la etiología y puede incluir terapia inmunomoduladora o antiinflamatoria, terapia modificadora de la enfermedad con agentes antifibróticos y, en casos avanzados, trasplante pulmonar. La atención médica debe ser holística, incluyendo rehabilitación pulmonar, prevención de infecciones y oxigenoterapia.
El objetivo de este artículo es realizar una actualización clínica de las enfermedades pulmonares intersticiales y comentar los avances en las estrategias diagnósticas y terapéuticas.
Interstitial lung diseases (ILDs) are a heterogeneous group of disorders that affect the lung interstitium, characterized by inflammation and/or fibrosis. Historically, they were based on histopathological characteristics of lung biopsies, but with the advancement of high-resolution computed tomography (HRCT), the approach has shifted, allowing for the identification of disease patterns that suggest different etiologies. The classification of ILDs has evolved, and they are currently classified according to etiology, histopathological and/or radiological patterns, and evolutionary course. Idiopathic interstitial pneumonias (IIPs) are a group of ILDs of unknown origin, with idiopathic pulmonary fibrosis (IPF) being the most common within the IIPs. The concept of progressive fibrosing interstitial lung disease (PF-ILD) has been developed, describing a phenotype of ILD with progression despite treatment. Diagnosis is based on a detailed clinical history, physical examination, pulmonary function tests, autoimmune serology, and imaging techniques such as HRCT. Treatment varies according to etiology and may include immunomodulatory or anti-inflammatory therapy, disease-modifying therapy with antifibrotic agents and, in advanced cases, lung transplantation. Medical care should be holistic, including pulmonary rehabilitation, infection prevention, and oxygen therapy. This article aims to provide a clinical update on interstitial lung diseases and comment on advances in diagnostic and therapeutic strategies.
Las enfermedades pulmonares intersticiales (EPI) constituyen un conjunto heterogéneo de trastornos que presentan una amplia variedad de etiologías y que se manifiestan clínica e imagenológicamente de manera similar1. Tradicionalmente agrupadas bajo la denominación inexacta de “fibrosis pulmonar”, las EPI se caracterizan por afectar el intersticio pulmonar con fenómenos inflamatorios y/o fibróticos2.
Históricamente, el diagnóstico y la clasificación de las EPI se basaban en las características histopatológicas observadas en las biopsias pulmonares3. Sin embargo, con el desarrollo y la mejora de la tomografía computarizada de alta resolución (TCAR) esto ha cambiado. La TCAR permite una visualización detallada de la estructura pulmonar y puede identificar diferentes patrones de enfermedad que sugieren etiologías4.
La histología, aunque sigue siendo una herramienta valiosa, ya no se considera el estándar de oro en el proceso diagnóstico y ha sido reemplazado por la opinión del comité multidisciplinario (CMD)5,6.
El CMD está compuesto por neumólogos especializados en EPI, radiólogos de tórax, patólogos pulmonares, reumatólogos, entre otros especialistas. Este enfoque colaborativo permite una discusión integral que conduce a una mayor precisión en el diagnóstico y a una mejor selección de las opciones terapéuticas7.
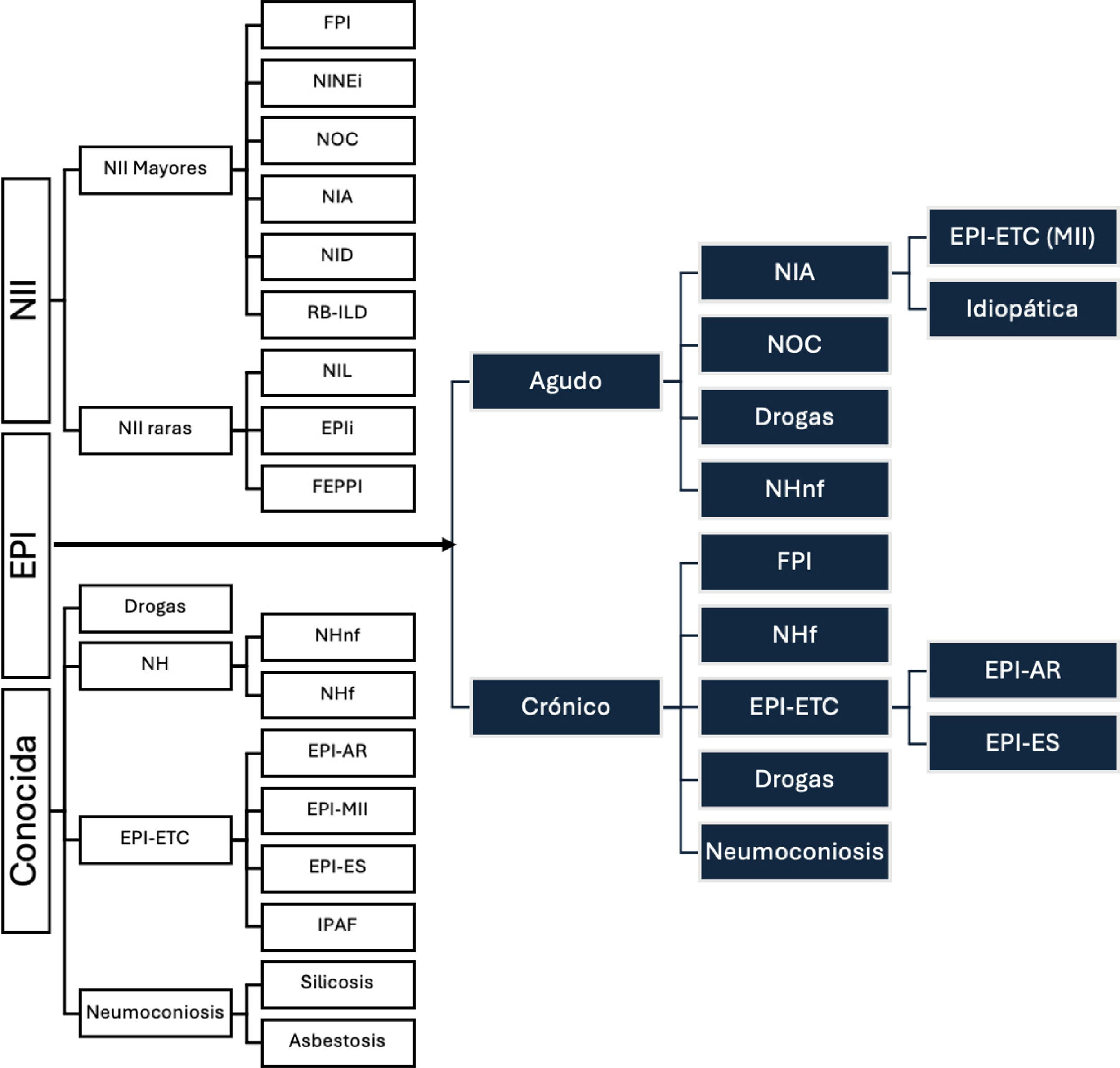
Clasificación y nuevos conceptosLa clasificación de las EPI ha experimentado una evolución significativa, reflejando una comprensión más profunda de los mecanismos patogénicos y el comportamiento clínico1. Las EPI se pueden clasificar desde múltiples perspectivas, incluyendo su etiología, patrones histopatológicos y/o radiológicos y un curso evolutivo agudo o crónico8. Figura 1.
8. EPI: enfermedad pulmonar intersticial; NII: neumonías intersticiales idiopáticas; FPI: fibrosis pulmonar idiopática; NINEi: neumonía intersticial no específica idiopática; NOC: neumonía en organización criptogénica; NIA: neumonía intersticial aguda; NID: neumonía intersticial descamativa, RB-ILD: bronquiolitis respiratoria con enfermedad pulmonar intersticial; NIL: neumonía intersticial linfocítica; EPIi: enfermedad pulmonar intersticial inclasificable; FEPPI: fibroelastosis pleuro-parenquimatosa idiopática; NH: neumonitis por hipersensibilidad; NHnf: neumonitis por hipersensibilidad no fibrótica; NHf: neumonitis por hipersensibilidad fibrótica; EPI-AR: EPI secundaria a artritis reumatoide; EPI-MII: EPI secundaria a miopatías inflamatorias; EPI-ES: EPI secundaria a esclerosis sistémica; IPAF: neumonía intersticial con rasgos de autoinmunidad (interstitial neumonia with autoimmune features).")
Clasificación de las enfermedades pulmonares intersticiales, según causa y evolución
Adaptado y modificado de: Travis WD et al. (2013)8.
EPI: enfermedad pulmonar intersticial; NII: neumonías intersticiales idiopáticas; FPI: fibrosis pulmonar idiopática; NINEi: neumonía intersticial no específica idiopática; NOC: neumonía en organización criptogénica; NIA: neumonía intersticial aguda; NID: neumonía intersticial descamativa, RB-ILD: bronquiolitis respiratoria con enfermedad pulmonar intersticial; NIL: neumonía intersticial linfocítica; EPIi: enfermedad pulmonar intersticial inclasificable; FEPPI: fibroelastosis pleuro-parenquimatosa idiopática; NH: neumonitis por hipersensibilidad; NHnf: neumonitis por hipersensibilidad no fibrótica; NHf: neumonitis por hipersensibilidad fibrótica; EPI-AR: EPI secundaria a artritis reumatoide; EPI-MII: EPI secundaria a miopatías inflamatorias; EPI-ES: EPI secundaria a esclerosis sistémica; IPAF: neumonía intersticial con rasgos de autoinmunidad (interstitial neumonia with autoimmune features).
Las neumonías intersticiales idiopáticas (NII): son un grupo de EPI de origen desconocido, en las que no se identifica un factor específico que contribuya a su desarrollo. La fibrosis pulmonar idiopática es la más común dentro de las NII, pero solo representa alrededor del 20% de todas las EPI, con frecuencias similares a la neumonitis por hipersensibilidad (NH) y las EPI asociadas a enfermedades del tejido conectivo (EPI-ETC). La clasificación vigente de las NII se actualizó en 2013; distinguiendo seis tipos principales y tres enfermedades raras8,9.
La fibrosis pulmonar idiopática (FPI) es la EPI más investigada, por definición es una enfermedad fibrosante progresiva y, en general tiene un pronóstico desfavorable10. Es más prevalente en hombres mayores de 65 años, especialmente entre aquellos con historial de tabaquismo. El diagnóstico de FPI se basa en la identificación de un patrón de neumonía intersticial usual (NIU) probable o típico en la TCAR junto con la exclusión de causas secundarias, como EPI-ETC, especialmente artritis reumatoide (AR) y vasculitis asociadas con anticuerpos contra el citoplasma de los neutrófilos (ANCA) (VAA), neumonitis por hipersensibilidad fibrótica (NHf), asbestosis, daño por drogas y sarcoidosis fibrótica11,12.
EPI de causa conocida: Este conjunto incluye las EPI derivadas de enfermedades del tejido conectivo (ETC), exposiciones a polvos orgánicos como la neumonitis por hipersensibilidad, exposiciones a polvos inorgánicos tales como la neumoconiosis, asbestosis y silicosis, y las EPI inducidas por fármacos, entre otras1,3,4.
Neumonitis por hipersensibilidad fibrótica (NHf): La NH es una EPI causada por una respuesta inmune inadecuada tras la inhalación de sustancias orgánicas, afectando a individuos previamente sensibilizados13. Esta respuesta inmune se caracteriza por la participación de IgG y linfocitos TH1, lo que resulta en una inflamación pulmonar con infiltrados linfocíticos y granulomas mal formados14. La neumonitis por hipersensibilidad (NH) se clasifica en formas no fibróticas (NHnf) predominando el compromiso inflamatorio con un curso clínico agudo o subagudo y fibróticas (NHf)15. La prevalencia es mayor en adultos mayores y varía según factores ambientales y culturales, siendo la forma fibrótica generalmente progresiva16.
EPI asociada a enfermedad del tejido conectivo (EPI-ETC): La EPI es una manifestación común de las ETC, impactando significativamente en la morbilidad y mortalidad de los pacientes17. La EPI puede ser diagnosticada al mismo tiempo que la ETC, manifestarse después de estas, o incluso aparecer antes. Entre el 15% y el 30% de los casos de EPI de causa desconocida podrían tener una ETC oculta. Además, algunos pacientes con EPI que no cumplen con los criterios formales para una ETC pueden mostrar signos de autoinmunidad clasificándose como un enfermedad intersticial con rasgos de autoinmunidad, (IPAF, por sus siglas en inglés de interstitial pneumonia with autoimmune features)18,19. Las ETC con mayor frecuencia de afectación pulmonar incluyen la esclerosis sistémica (ES) 50-60%, la AR 10-30% y las miopatías inflamatorias idiopáticas (MII) 20%-86%20–22. Dentro de las MII, el síndrome antisintetasa (SAS) es un subgrupo específico caracterizado por artritis, miositis y un anticuerpo antisintetasa (ac-AS). En este grupo, la EPI es altamente frecuente (70-100%), dependiendo del anticuerpo específico23,24.
Recientemente, se ha desarrollado el concepto de enfermedad pulmonar intersticial fibrosante progresiva (EPI-FP)25. Este describe un fenotipo de EPI que, independientemente de su etiología, muestra progresión de la enfermedad a pesar del tratamiento específico26. Tabla 1. Es fundamental diferenciar este concepto de la enfermedad pulmonar intersticial fibrótica (EPI-F), que se refiere a la EPI donde predominan los elementos fibróticos tanto en la TCAR como en la histopatología. Esta distinción subraya que la EPI-FP es un “fenotipo de comportamiento” y no una entidad clínica independiente26–29. Figura 2.
Enfermedad pulmonar intersticial fibrosante progresiva (EPI-FP): criterios, clínicos, imagenológicos y funcionales
| Empeoramiento de los síntomas respiratorios | |
| Evidencia fisiológica de progresión de la enfermedad (cualquiera de los siguientes): | |
| a. | Disminución absoluta en CVF ≥5% del teórico en 12 meses de seguimiento. |
| b. | Disminución absoluta en DLCO ≥10% del teórico en 12 meses de seguimiento. |
| Evidencia radiológica de progresión de la enfermedad (uno o más de los siguientes): | |
| a. | Aumento en la extensión o severidad de las bronquiectasias o bronquiolectasias por tracción |
| b. | Nueva opacidad en vidrio esmerilado con bronquiectasia por tracción |
| c. | Nuevas reticulaciones finas |
| d. | Aumento en la extensión o el grosor de las anomalías reticulares |
| e. | Incremento o nuevas zonas con panal abeja |
| f. | Aumento en la reducción de volúmenes pulmonares |
Adaptado y modificado de Rajan SK et al. (2023)26.
Abreviaturas: DLCO: prueba de difusión de monóxido de carbono; CVF: capacidad vital forzada.
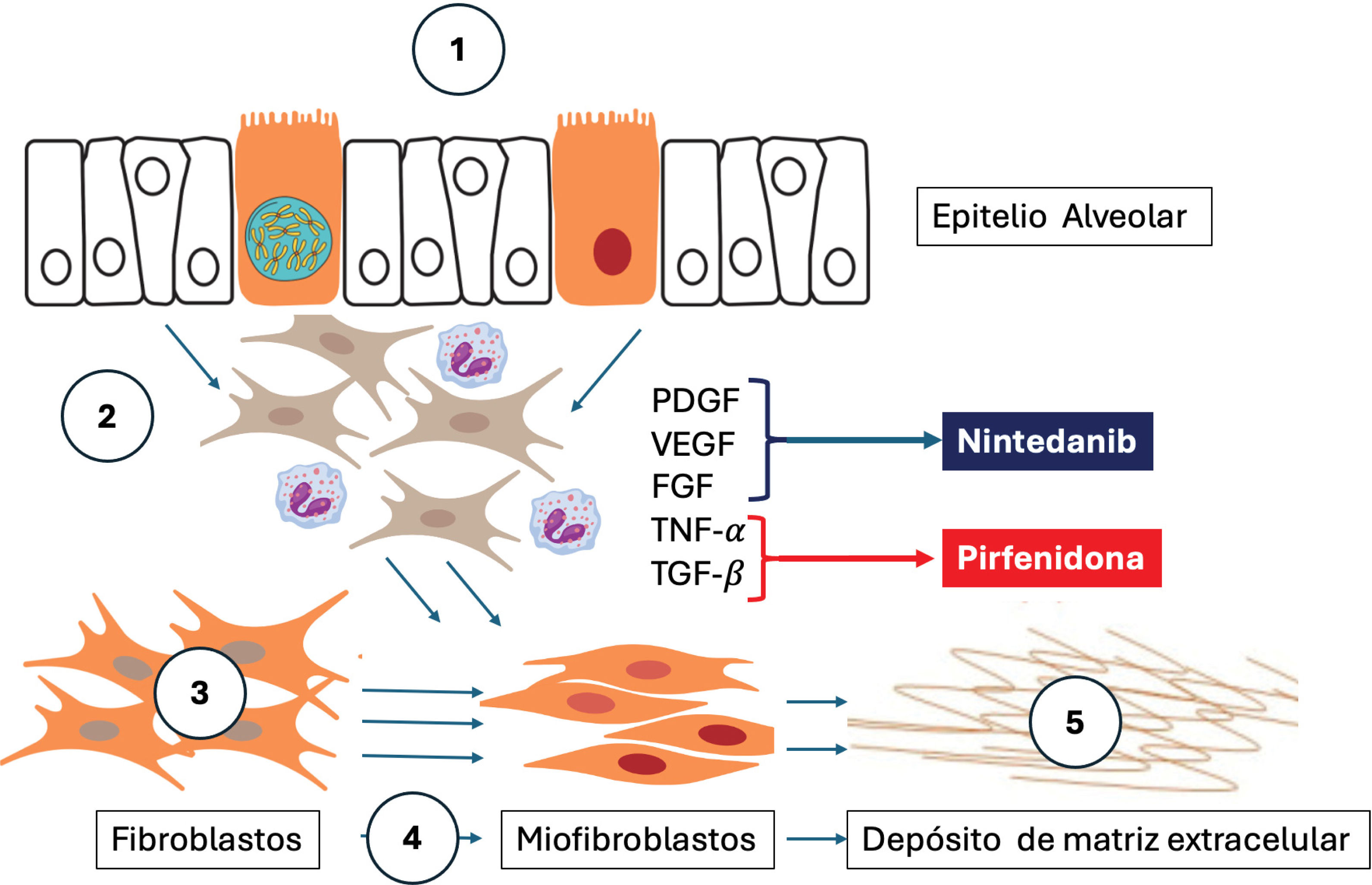
 y blancos de acción de la pirfenidona y el nintedanib Adaptado de Shumar JN et al. (2021)30 y Spagnolo P et al (2021)31. 1. Epitelio alveolar donde ocurren mecanismos genéticos y epigenéticos (mutaciones que afectan la longitud telomérica, la síntesis de surfactante pulmonar y la mucina; y metilaciones del DNA, modificaciones de histonas y RNA no codificante) inducidos por estímulos patogénicos reiterados como infecciones respiratorias, reflujo gastroesofágico (RGE), tabaquismo, polución y la inhalación de sustancias nocivas. 2. La respuesta inflamatoria inicial implica la migración de fibroblastos, linfocitos TH1 y TH2 y neutrófilos. En estados anormales, se liberan citocinas inflamatorias como el factor de crecimiento derivado de plaquetas (PDGF), el factor de crecimiento endotelial vascular (VEGF), el factor de crecimiento de fibroblastos (FGF), el factor de necrosis tumoral alfa (TNF-α) y el factor de crecimiento transformante beta (TGF-β), que son dianas para los tratamientos antifibróticos como nintedanib y pirfenidona. 3. Proliferación de fibroblastos. 4. Transformación descontrolada de fibroblastos en miofibroblastos con pérdida de mecanismos de apoptosis. 5. Depósito excesivo de matriz extracelular y fibrosis28,29.")
Fisiopatología de la enfermedad pulmonar intersticial (EPI) y blancos de acción de la pirfenidona y el nintedanib
Adaptado de Shumar JN et al. (2021)30 y Spagnolo P et al (2021)31.
1. Epitelio alveolar donde ocurren mecanismos genéticos y epigenéticos (mutaciones que afectan la longitud telomérica, la síntesis de surfactante pulmonar y la mucina; y metilaciones del DNA, modificaciones de histonas y RNA no codificante) inducidos por estímulos patogénicos reiterados como infecciones respiratorias, reflujo gastroesofágico (RGE), tabaquismo, polución y la inhalación de sustancias nocivas.
2. La respuesta inflamatoria inicial implica la migración de fibroblastos, linfocitos TH1 y TH2 y neutrófilos. En estados anormales, se liberan citocinas inflamatorias como el factor de crecimiento derivado de plaquetas (PDGF), el factor de crecimiento endotelial vascular (VEGF), el factor de crecimiento de fibroblastos (FGF), el factor de necrosis tumoral alfa (TNF-α) y el factor de crecimiento transformante beta (TGF-β), que son dianas para los tratamientos antifibróticos como nintedanib y pirfenidona.
3. Proliferación de fibroblastos.
4. Transformación descontrolada de fibroblastos en miofibroblastos con pérdida de mecanismos de apoptosis.
5. Depósito excesivo de matriz extracelular y fibrosis28,29.
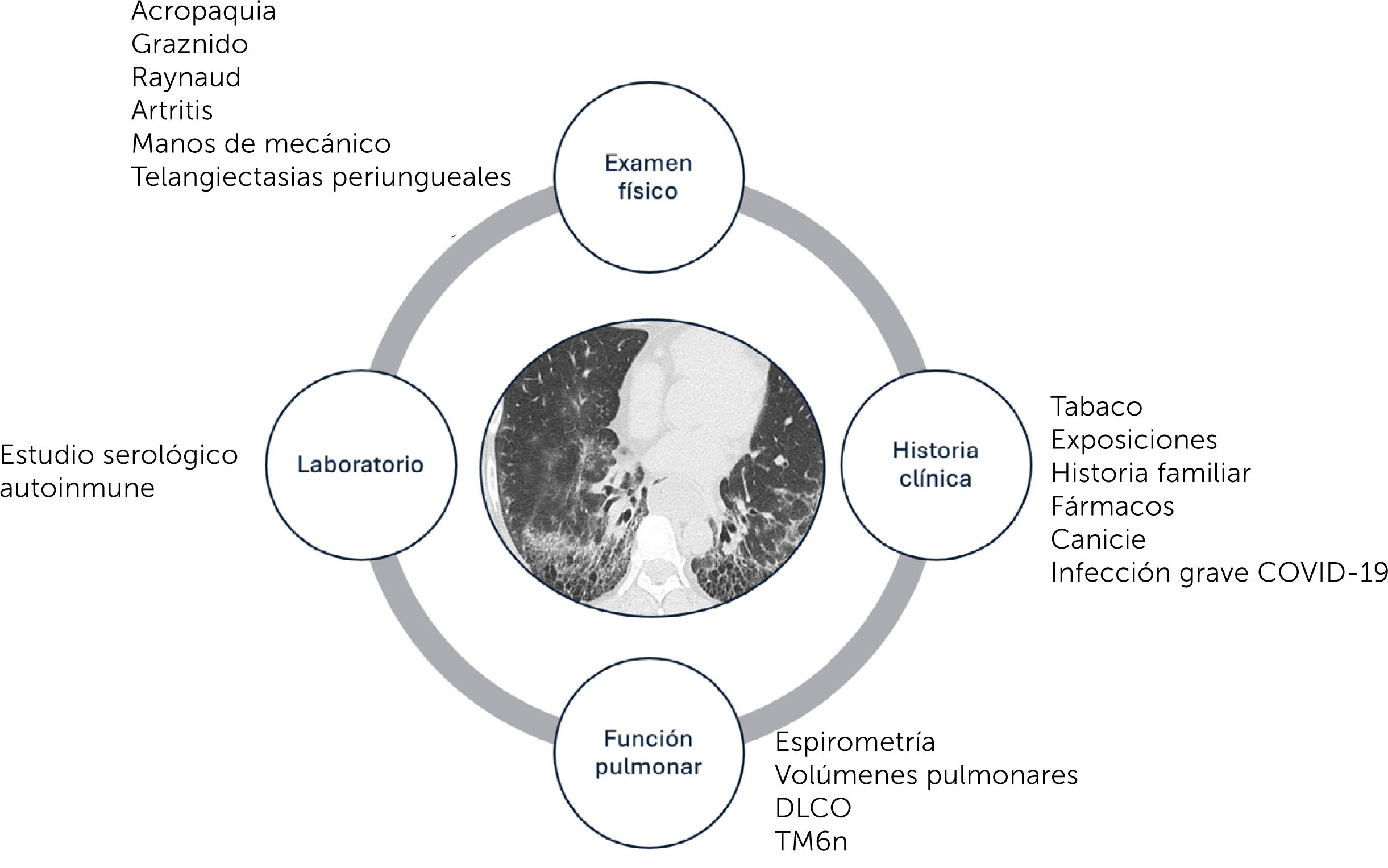
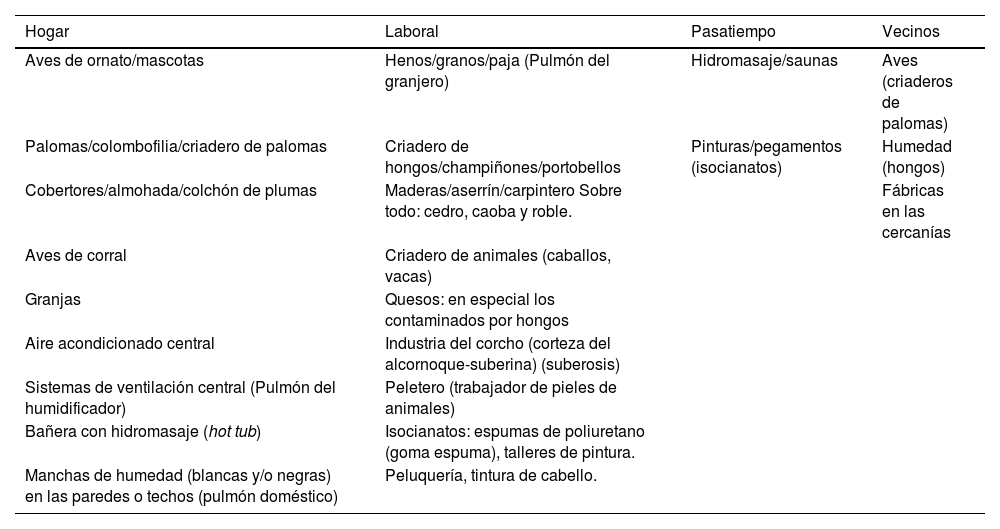
La anamnesis debe incluir datos demográficos como edad, sexo y etnia, antecedentes familiares de EPI, tabaquismo, exposiciones, ETC prexistente y antecedentes de infecciones graves por COVID-19. Se debe registrar la agregación familiar de casos (hermanos y padres) que podría sugerir la posibilidad de fibrosis pulmonar familiar1,3,6. Una historia detallada de exposiciones ambientales y ocupacionales, incluyendo el contacto con aves y exposición a ambientes húmedos o mohosos permite dirigir la sospecha diagnóstica a NH15,16. Herramientas como encuestas de exposición pueden ser útiles para identificar exposiciones no evidentes (tabla 2). Se deben revisar los fármacos o drogas que se sospechen puedan tener rol patogénico (www.pneumotox.com/)30,31.
Exposiciones más frecuentes, relacionadas con neumonitis por hipersensibilidad
| Hogar | Laboral | Pasatiempo | Vecinos |
|---|---|---|---|
| Aves de ornato/mascotas | Henos/granos/paja (Pulmón del granjero) | Hidromasaje/saunas | Aves (criaderos de palomas) |
| Palomas/colombofilia/criadero de palomas | Criadero de hongos/champiñones/portobellos | Pinturas/pegamentos (isocianatos) | Humedad (hongos) |
| Cobertores/almohada/colchón de plumas | Maderas/aserrín/carpintero Sobre todo: cedro, caoba y roble. | Fábricas en las cercanías | |
| Aves de corral | Criadero de animales (caballos, vacas) | ||
| Granjas | Quesos: en especial los contaminados por hongos | ||
| Aire acondicionado central | Industria del corcho (corteza del alcornoque-suberina) (suberosis) | ||
| Sistemas de ventilación central (Pulmón del humidificador) | Peletero (trabajador de pieles de animales) | ||
| Bañera con hidromasaje (hot tub) | Isocianatos: espumas de poliuretano (goma espuma), talleres de pintura. | ||
| Manchas de humedad (blancas y/o negras) en las paredes o techos (pulmón doméstico) | Peluquería, tintura de cabello. |
Adaptado de la historia clínica utilizada en el consultorio EPID del Hospital de Rehabilitación Respiratoria “María Ferrer”, Buenos Aires, Argentina.
Los síntomas y signos de la EPI son inespecíficos, siendo la disnea en actividad la más común. Las crepitaciones son hallazgos característicos, mientras que otros ruidos respiratorios, como el graznido, pueden indicar compromiso de la vía aérea pequeña y orientar hacia diagnósticos específicos como la NH, EPI-AR o EPI-Sindrome de Sjögren (SS)32. La acropaquía, más común en la FPI pero también presente en otras EPI en etapas avanzadas, y signos sutiles como la presencia de canicie precoz antes de los de los 40 años, orienta a EPI familiar. Otros elementos clínicos sugerentes de EPI-ETC19–21 se describen en la tabla 3.
Síntomas y signos más frecuentes según enfermedades del tejido conectivo
| Hogar | Laboral |
|---|---|
| Artritis reumatoide | Poliartralgias/ArtritisRigidez matinal (>30 min)Poli serositis |
| Esclerosis sistémica | Fenómeno de RaynaudTelangiectasias cuadradas (cara o mucosas)EsclerodactiliaPitting scar/Úlceras digitalesTelangiectasias periunguealesReflujo gastroesofágico |
| Miopatías inflamatorias | DisfagiaDebilidad muscularPuffy hands (edema de manos)Manos de mecánicoHiker's feet (pies de excursionista)GottronErythema in shawl (eritema en V o Chal)Erythema in holsters (eritema en pistoleras)Eritema heliotropo |
| Otras | Xeroftalmia/XerostomíaÚlceras oralesFotosensibilidadPérdida de peso (>10%) |
| Estudios complementarios | Biopsia de glándulas salivales menoresCapilaroscopia del lecho ungueal |
La tomografía computarizada de alta (TCAR) es una herramienta diagnóstica esencial, permitiendo no solo la identificación de patrones fibróticos y no fibróticos, sino también proporcionando información valiosa sobre el pronóstico.
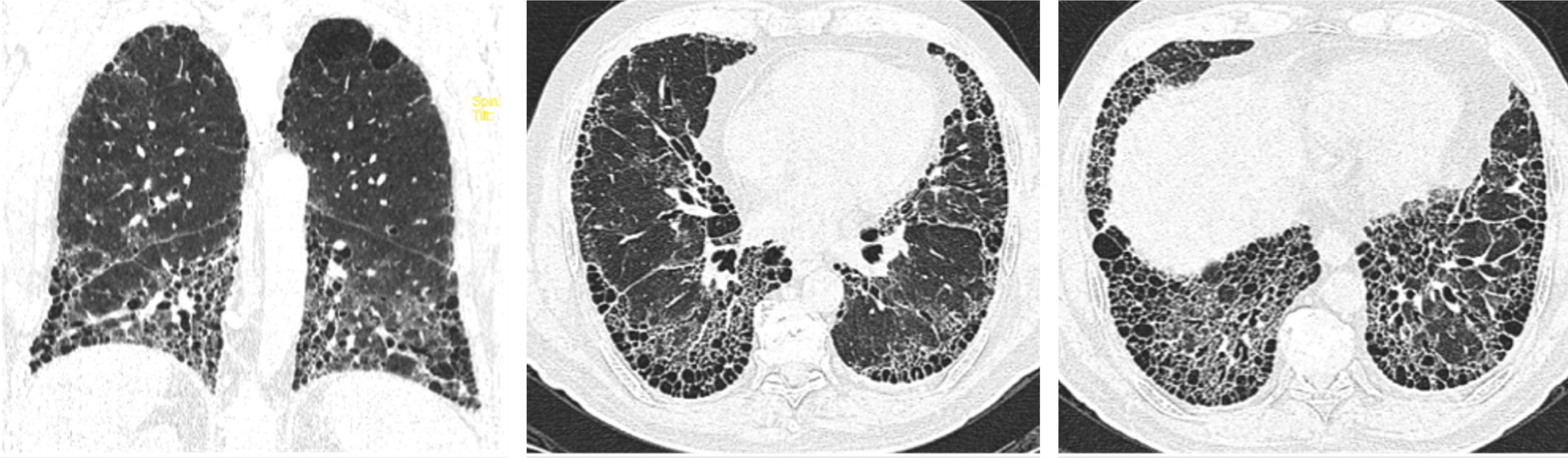
Neumonía intersticial usual (NIU)Este patrón se distingue por reticulado intersticial con orientación apicocaudal y subpleural, bronquiectasias por tracción y, de manera distintiva para el patrón típico frente al probable, la presencia de panal de abeja o honeycombing1,3,4,8,33. (Figura 3).
 con patrón de neumonía intersticial usual (NIU) típico .")
Aunque es patognomónico de la FPI, también puede observarse en NHf, EPI-AR, EPI-ES, daño por drogas, asbestosis y sarcoidosis fibrótica entre otras.
Algunos signos como la presencia de panal de abeja anterior o “cuatro esquinas”, panal exuberante, y esófago dilatado, pueden sugerir un patrón NIU secundario a ETC.
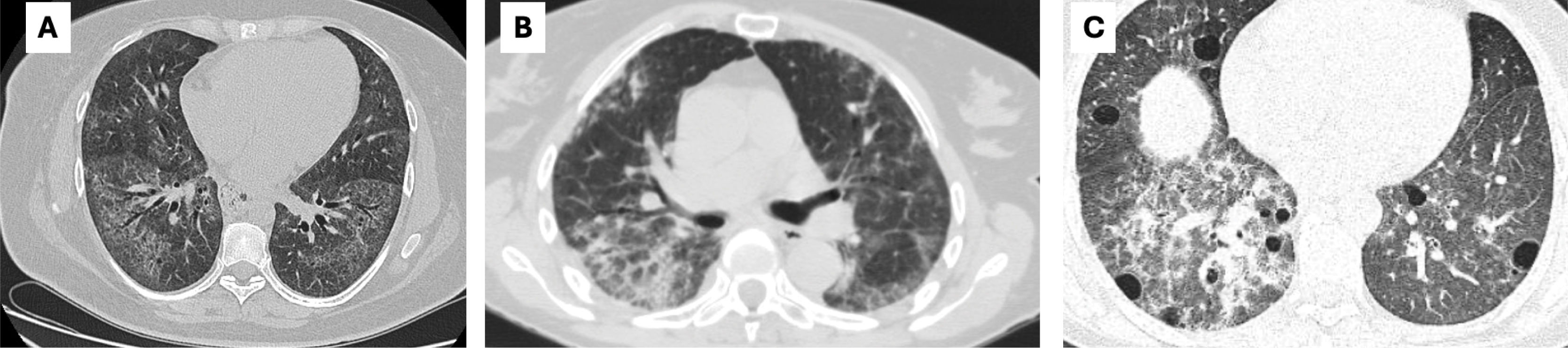
Neumonitis intersticial no específica (NINE).Figura 4A: Caracterizada por opacidades en vidrio esmerilado (VE) y un patrón reticular difuso en las bases, puede incluir bronquiectasias por tracción en la NINE fibrótica o predominar el VE en la NINE celular8,33,34. El respeto subpleural es el elemento más específico. Este patrón se asocia frecuentemente a ETC, especialmente a la ES.
 con patrón de neumonía intersticial no específica (NINE), B: TCAR con patrón de neumonitis intersticial no específica con patrón de neumonía en organización (NINE-NO) y C: TCAR con patrón de neumonía intersticial linfocítica (NIL).")
Patrones tomográficos alternativos en neumonía intersticial usual.
A: Tomografía computarizada de alta (TCAR) con patrón de neumonía intersticial no específica (NINE), B: TCAR con patrón de neumonitis intersticial no específica con patrón de neumonía en organización (NINE-NO) y C: TCAR con patrón de neumonía intersticial linfocítica (NIL).
Neumonía en organización (NO).Figura 4B: Presenta opacidades alveolares en VE o condensantes, migratorias en parches de distribución subpleurales o peribroncovasculares1,8. Este patrón puede encontrarse en diversas condiciones, incluyendo reacciones a drogas, etc, son más frecuente en las MII o en su forma idiopática NOC (neumonía en organización criptogénica).
NINE-NO (neumonitis intersticial no específica con patrón de neumonía en organización): Integra características de NINE y NO, siendo este patrón altamente indicativo de ETC, particularmente en subtipos como el SAS1,8,33,34.
Neumonía intersticial linfocítica (NIL).Figura 4C: Se manifiesta por opacidades bilaterales y difusas en vidrio esmerilado, nódulos centrolobulillares mal definidos, nódulos subpleurales, engrosamiento peribroncovascular y la presencia de espacios aéreos quísticos de paredes finas con distribución aleatoria. Este patrón es específico de ETC, como el SS8. Su forma idiopática (NILi), es clasificada como una EPI rara en las NII.
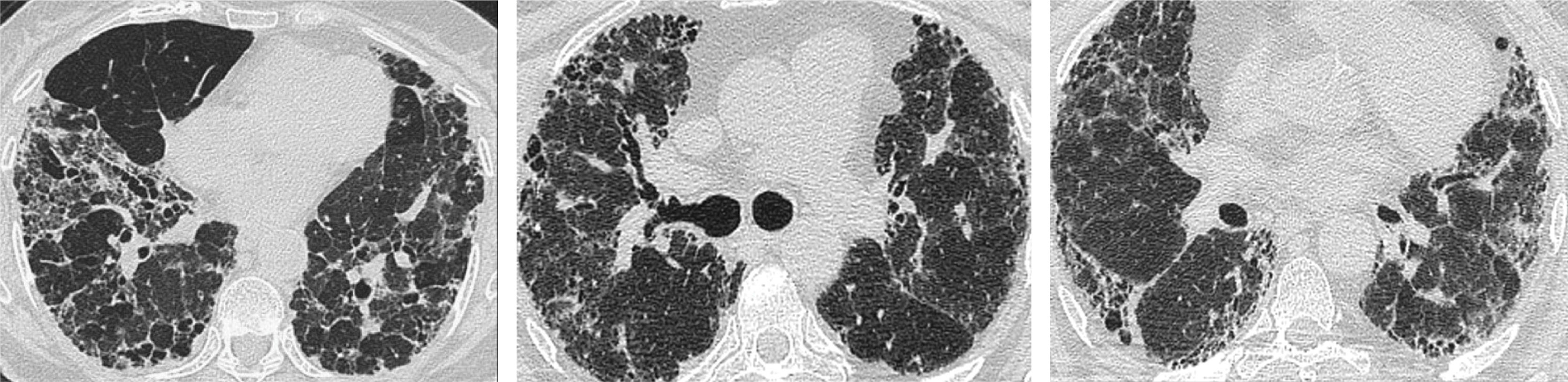
Patrón de neumonitis por hipersensibilidad fibrótico (NHf)Combina elementos fibróticos y de compromiso de la vía aérea pequeña. En la figura 5 se observa reticulación intersticial gruesa con distorsión de la arquitectura pulmonar, predominante axial, con una distribución que suele ser desde los lóbulos superiores o zonas media y relativamente preservada en las zonas inferiores15,16. La bronquiectasia por tracción y el panal de abeja pueden estar presentes pero no dominan. Las anomalías que indican enfermedad de la vía aérea pequeña incluyen nódulos centrolobulillares mal definidos y/u opacidades en vidrio esmerilado, atenuación en mosaico, patrón de tres densidades y/o atrapamiento aéreo, frecuentemente en una distribución lobulillar33,35,36. Figura 6.
 con patrón de neumonitis por hipersensibilidad fibrótica (NHf).")
 y patrón radiológico. Patrón de neumonía intersticial usual (NIU), el objetivo diagnóstico debería ser excluir causas secundarias, considerando la FPI como diagnóstico más probable. Patrones como la neumonitis intersticial no específica (NINE) o NINE/NO (neumonía en organización) búsqueda de enfermedad de tejido conectivo oculta. Patrón de neumonitis por hipersensibilidad (NH) de manera compatible o típica, búsqueda dirigida del antígeno causante. DLCO: capacidad de difusión del monóxido de carbono; TM6m: el test de marcha de 6 minutos.")
Esquema diagnóstico con enfoque en la tomografía computarizada de alta resolución (TCAR) y patrón radiológico.
Patrón de neumonía intersticial usual (NIU), el objetivo diagnóstico debería ser excluir causas secundarias, considerando la FPI como diagnóstico más probable. Patrones como la neumonitis intersticial no específica (NINE) o NINE/NO (neumonía en organización) búsqueda de enfermedad de tejido conectivo oculta.
Patrón de neumonitis por hipersensibilidad (NH) de manera compatible o típica, búsqueda dirigida del antígeno causante.
DLCO: capacidad de difusión del monóxido de carbono; TM6m: el test de marcha de 6 minutos.
- Las pruebas de función pulmonar (PFP): la espirometría, la capacidad de difusión del monóxido de carbono (DLCO, por sus siglas en inglés), los volúmenes pulmonares y el test de marcha de 6 minutos (TM6m), son las PFP fundamentales aunque no diagnostican directamente la EPI ni distinguen su causa, permiten definir severidad, pronóstico, seguimiento y respuesta a terapia1,4,9. El TM6m mide la capacidad de ejercicio submáximo, reflejando la tolerancia al ejercicio, la necesidad de oxígeno suplementario en actividad y la respuesta a la rehabilitación pulmonar37,38.
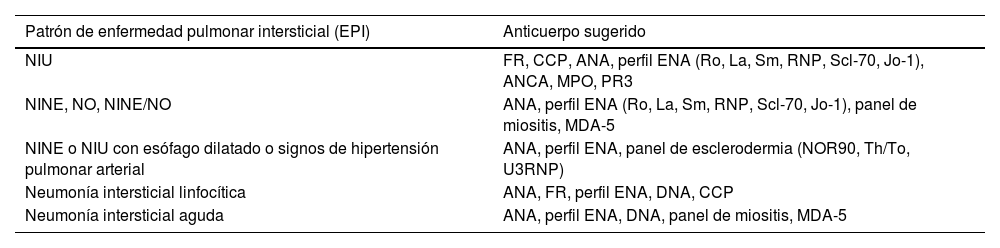
- La serología autoinmune desempeña un papel esencial en el diagnóstico de la EPI-ETC; sin embargo, su interpretación puede ser compleja, el rol patogénico de un anticuerpo positivo, debe correlacionarse con la clínica y otros estudios complementarios39,40. (Tabla 4).
Patrones en la tomografía computarizada de alta y estudio serológico recomendado según sospecha de enfermedad del tejido conectivo
| Patrón de enfermedad pulmonar intersticial (EPI) | Anticuerpo sugerido |
|---|---|
| NIU | FR, CCP, ANA, perfil ENA (Ro, La, Sm, RNP, Scl-70, Jo-1), ANCA, MPO, PR3 |
| NINE, NO, NINE/NO | ANA, perfil ENA (Ro, La, Sm, RNP, Scl-70, Jo-1), panel de miositis, MDA-5 |
| NINE o NIU con esófago dilatado o signos de hipertensión pulmonar arterial | ANA, perfil ENA, panel de esclerodermia (NOR90, Th/To, U3RNP) |
| Neumonía intersticial linfocítica | ANA, FR, perfil ENA, DNA, CCP |
| Neumonía intersticial aguda | ANA, perfil ENA, DNA, panel de miositis, MDA-5 |
Adaptado y modificado de Wolff y Valenzuela (2019)39.
Abreviaturas: NIU: neumonía intersticial usual; NINE: neumonía intersticial no específica; NO: neumonía en organización; FR: factor reumatoide; CCP: péptidos citrulinados cíclicos; ANA: anticuerpos antinucleares; ENA: antígenos nucleares extraíbles; ANCA: anticuerpos anticitoplasma de neutrófilos; MDA-5: melanoma differentiation-associated protein 5; MPO: anticuerpo antimieloperoxidasa; PR3: anticuerpo antiproteinasa 3.
Los anticuerpos antinucleares (ANA) son los autoanticuerpos más frecuentemente solicitados19,20. La detección se realiza mediante inmunofluorescencia indirecta sobre células HEp-2 que identifica la fluorescencia específica de distintos antígenos presentes en el núcleo celular. Los resultados se informan con un título, menor a 1/80, generalmente se considera negativo; los títulos superiores se reportan como positivos acompañados de un patrón específico, ya sea nucleolar, homogéneo, moteado o centromérico.
La presencia de ANA no necesariamente indica una enfermedad; por ejemplo, hasta un 20% de las mujeres sanas pueden tener un ANA positivo en títulos bajos (1/80 a 1/160)19,23. Sin embargo, títulos más elevados suelen ser patológicos, asociándose principalmente con enfermedades autoinmunes, procesos infecciosos y neoplásicos. Los cambios recientes en la clasificación del consenso internacional sobre patrones de ANA (ICAP, por sus siglas en inglés) han refinado esta interpretación, solicitando la notificación no solo de la presencia de fluorescencia nuclear, sino también citoplasmática y del aparato mitótico (https://www.anapatterns.org). Esto es relevante para autoanticuerpos como los ac-AS, cuyos antígenos se encuentran en el citoplasma y podrían explicar hasta el 60% de los ANA negativos en SAS. La inclusión de otros autoanticuerpos, como el perfil de antígenos nucleares extraíbles (ENA, por sus siglas en inglés de extractable nuclear antigens), que típicamente incluye la detección de anti-Sm (Smith), anti-RNP (ribonucleoproteína nuclear U1), anti-SS-A/Ro (antígeno A relacionado con el SS o proteína Ro) y anti-SS-B/La (antígeno B relacionado con el SS o proteína La), anti-Scl-70 (topoisomerasa I) y anti-Jo-1 (histidil-ARNt sintetasa-único ac-AS descrito en el perfil ENA). Los anticuerpos anti-péptido citrulinado cíclico (anti-CCP) y el factor reumatoide (FR) también deben formar parte del estudio inmunológico inicial30.
Un estudio serológico más específico se debe plantear cuando existe alta sospecha de una ETC oculta. El panel para miopatías identifica anticuerpos específicos de miopatía, entre ellos los ac-AS (Jo-1, PL-12 (alanil-ARNt sintetasa), PL-7 (treonil-ARNt sintetasa), EJ (glicina-ARNt sintetasa) y OJ (isoleucil-ARNt sintetasa) que son excluyentes entre sí y se encuentran en aproximadamente el 25-30% de los casos de MII22–24. Además identifica anticuerpos relacionados como Ro52/TRIM21, PM/Scl (polimiositis/esclerodermia) y Ku (subunidad reguladora ADN-PK) o MDA5 (melanoma differentation-associated protein 5) anticuerpo asociado a una EPI rápidamente progresiva (EPI-RP) y de curso agudo y grave41. El panel de esclerodermia es otra herramienta menos común pero mucho más específica que principalmente nos proporciona la identificación de anticuerpos como NOR90/hUBF (región 90 organizadora del nucléolo/factor de unión humano río arriba), Th/To (ribonucleasas mitocondrial y P) y U3-RNP (anti-fibrilarina), los cuales son marcadores específicos en ES y sobre todo en la forma “sine” donde predomina el compromiso pulmonar con escasas manifestaciones extrapulmonares. Recientemente se ha establecido que la VAA también podría manifestarse como EPI22,42,43. Se recomienda solicitar pruebas de ANCA (ANCA-p, ANCA-c), MPO (mieloperoxidasa) y PR-3 (proteinasa 3), especialmente ante TCAR con patrón NIU y elementos clínicos o de laboratorio sospechosos.
Estudios invasivos e histológicos: La fibrobroncoscopia (FBC) y el lavado broncoalveolar (LBA) presentan una utilidad limitada en casos donde se identifica un patrón de NIU en la TCAR. Generalmente, no se aconseja su realización salvo que existan indicios de procesos infecciosos o neoplásicos1,4,8. El LBA puede ser de utilidad diagnóstica en otras EPI tales como la NH caracterizada por un recuento celular con predominio linfocítico; la sarcoidosis, que se distingue por un incremento en la relación CD4/CD8 de células T; la NINE celular, con linfocitosis; la neumonía intersticial aguda (NIA) asociada a un predominio de neutrófilos; la neumonía eosinofílica (NE), que cursa con eosinofilia, y la hemorragia alveolar difusa (HAD)44,45. La biopsia transbronquial (BTB) convencional desempeña un papel limitado en el diagnóstico, aunque puede contribuir en la identificación de NH, NO, NE, sarcoidosis y linfangitis carcinomatosa. En los últimos años, la BTB mediante el uso de criosondas, conocida como criobiopsia, ha evidenciado una mayor eficacia diagnóstica, llegando a ser comparable con la biopsia quirúrgica en términos de rendimiento. Sin embargo, esta técnica no está exenta de riesgos y puede conllevar complicaciones como neumotórax y hemoptisis46,47. La biopsia pulmonar quirúrgica se considera el método de referencia, pero implica riesgos considerables, incluyendo una tasa de mortalidad del 1,7%. Este porcentaje puede incrementarse significativamente en aquellos pacientes que están hospitalizados por insuficiencia respiratoria, observándose riesgo elevado de una exacerbación1,4,48.
En pacientes con signos clínicos y radiológicos definidos que sugieren un diagnóstico de FPI o aquellos con ETC manifiestas o exposiciones conocidas con sospecha de NH, generalmente no se requiere realizar una biopsia pulmonar. El CMD desempeña un papel crucial en la evaluación y la toma de decisiones de realizar biopsia u otros estudios complementarios5–7.
TratamientoEs crucial identificar comorbilidades que pueden impactar significativamente en la calidad de vida antes de comenzar un tratamiento. Condiciones como depresión, síndrome de apnea obstructiva del sueño (SAOS), reflujo gastroesofágico (RGE) e hipertensión arterial pulmonar (HAP)37,49.
Terapia no farmacológicaLa rehabilitación pulmonar: es el pilar más importante de la terapia, ya que es la única intervención que ha demostrado mejorar la capacidad de ejercicio, la autonomía del paciente y, en general, la calidad de vida. Además, formar parte de un equipo junto con otros pacientes que experimentan la misma enfermedad facilita la empatía respecto a las experiencias compartidas y ayuda a mejorar el estado de ánimo37,38,49.
La prevención de infecciones: en todos los pacientes, no solo en aquellos con inmunosupresión, se debe realizar vacunación contra el neumococo, la influenza anual y el COVID-19.
La oxigenoterapia: los programas de oxigenoterapia domiciliaria están diseñados para pacientes con EPOC. Sin embargo, para la EPI, no hay indicaciones claras ni una evidencia específica que demuestre los beneficios de la oxigenoterapia50,51. En la práctica, cuando existe hipoxemia en reposo, se indica oxigenoterapia domiciliaria continua, aumentando los flujos basales para actividades que requieren esfuerzo52,53. En casos donde solo se presenta hipoxemia inducida por el ejercicio, se prefiere el soporte con oxígeno durante dicha actividad y, si es posible, también de forma nocturna, siempre que se pueda constatar la caída de la saturación nocturna mediante oximetría o poligrafía53.
Cuidados paliativos: en etapas avanzadas de la enfermedad, al igual que en las enfermedades oncológicas, los cuidados paliativos buscan manejar los síntomas como la tos y la disnea que suelen ser muy incapacitantes1,4,54.
Terapia farmacológicaLa elección del tratamiento debe basarse en la etiología de la EPI, patrón radiológico, comportamiento clínico y funcional y, no menos importante, el performance del paciente55.
Terapia inmunomoduladora o antiinflamatoriaLos corticosteroides, el micofenolato de mofetilo (MMF), la azatioprina, la ciclofosfamida (CYC) y el rituximab son los fármacos más utilizados. La indicación se considera cuando hay signos de un proceso inflamatorio activo que puede ser tratado o la presencia de una enfermedad sistémica asociada, como la EPI-ETC20, que requiere de una terapia inmunosupresora. Esta terapia no solo se dirige a tratar la afección pulmonar sino también a los síntomas extrapulmonares y a modular el sistema inmune21,22.
En la neumonitis por hipersensibilidad, está claro que el tratamiento con esteroides es útil en su forma no fibrótica. En la NHf no existe evidencia y extrapolamos su tratamiento de otras EPI56, la presencia de graznido, exposición conocida identificada, atrapamiento aéreo, vidrio esmerilado o tres densidades en el TCAR, elementos de atrapamiento aéreo en las PFP, podrían sugerir algún grado de actividad inflamatoria y justificar el inicio de terapia inmunomoduladora57. La azatioprina y el MMF asociados a un corticosteroide como la prednisona han sido los más usados. Ambas terapias tienen un rol limitado y han demostrado estabilizar la caída de la función pulmonar. Ambos fármacos parecen ser igual de efectivos, con un perfil de tolerancia y seguridad ligeramente mejor para el micofenolato de mofetilo58.
En el manejo de la EPI-ETC la decisión es determinar qué pacientes requieren terapia específica para la EPI y a cuáles solo se le debe realizar seguimiento. Dada la escasez de evidencia directa, las recomendaciones actuales se basan en la opinión de expertos y en evidencia indirecta20. Es esencial considerar el tratamiento farmacológico para pacientes con enfermedad grave, extensión en la TCAR mayor a un 20%, elementos radiológicos de actividad como VE, condensaciones, PFP con capacidad vital forzada (CVF) menos del 70%, DLCO menos 80%, TM6m con desaturación, progresión al comparar PFP anteriores o factores de mal pronóstico específicos para cada condición21,22,59. Tabla 560–63.
EPI-ETC específicas, factores de riesgo y opciones terapéuticas
| EPI | Factores de riesgo | Opciones de tratamiento |
|---|---|---|
| EPI-ES | Anti-Scl-70, sexo masculino, raza negra, >modified Rodnan skin score (mRSS)* | MMF o CyC (cualquiera de las dos opciones puede usase como primera línea)59,60, rituximab61,62 como primera línea asociado o no a MMF o como terapia para EPI refractaria. Tocilizumab, casos seleccionados (parámetros inflamatorios elevados, EPI inflamatoria no extensa |
| EPI-AR | Hombre, anti-CCP títulos altos, Tabaquismo, patrón NIU | Rituximab MMF o CyC (en combinación con terapia para tratar manifestaciones articulares DMARD, otras terapias anti-TNF (infliximab, adalimumab, etanercept), abatacept y tofacitinib |
| EPI-MII | Anti-Jo1, anti-PL-7, anti-PL-12, raza negra | Corticosteroides en dosis altas asociado a azatioprina, MMF, CyC, rituximab o tofacitinib |
| EPI-RP (MDA5, SAS) | - | Considerar la terapia combinada anticalcineurínicos como la ciclosporina o el tacrolimus, junto con rituximab o CyC, dosis altas de corticoesteroides y la administración de inmunoglobulina intravenosa |
| Otros | - | Según el patrón radiológico dominante. Si es NINE, tratar como EPI-ES, si el subtipo dominante es NO, tratar como EPI-MII, si el subtipo dominante es NIU, es discutible el uso de terapia con nintedanib o pirfenidona asociada a IS o solo terapia IS, estos casos deben evaluarse según cada paciente. |
Adaptado y modificado de (Maher TM y Wuyts W (2019)59.
EPI-ES: enfermedad pulmonar intersticial-esclerosis sistémica; EPI-AR: enfermedad pulmonar intersticial-artritis reumatoide; EPI-MII: enfermedad pulmonar intersticial-miopatías inflamatorias idiopáticas; IPAF: neumonía intersticial con características autoinmunes; NIA: neumonia intersticial aguda; EPI-RP: EPI rápidamente progresiva; Ac-AS: anticuerpos antisintetasa; MDA5: melanoma differentiation-associated protein 5; IS: inmunosupresión; MMF: micofenolato de mofetilo; CyC: ciclofosfamida; DMARD: fármacos modificadores de la enfermedad antirreumática; TNF: factor de necrosis tumoral; NINE: neumonía intersticial no específica; NO: neumonía organizada; NIU: neumonía intersticial usual.
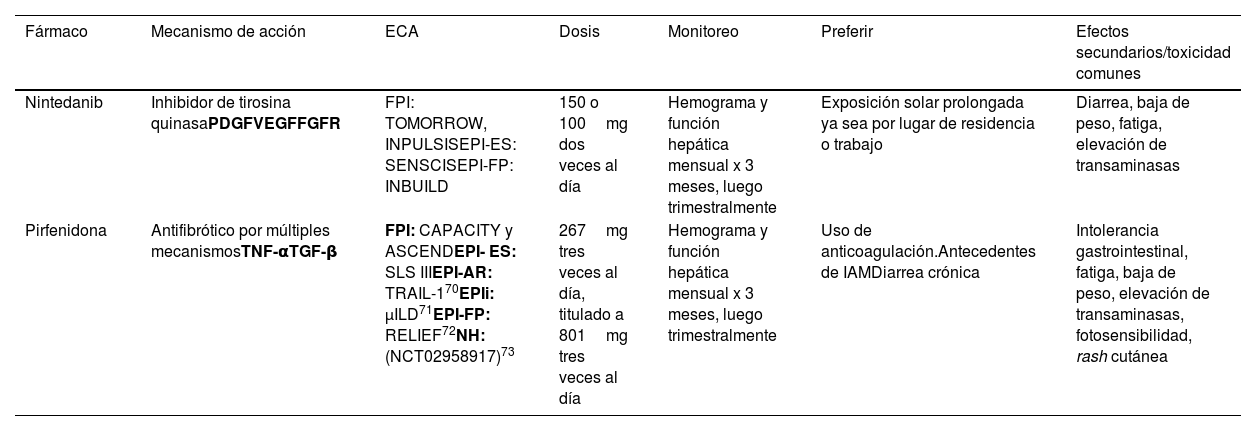
En pacientes con FPI y EPI-FP, se recomienda el uso de la terapia antifibrótica. En el año 2014, tres grandes ensayos aleatorios controlados con placebo demostraron una disminución más lenta de la CVF en pacientes con FPI tratados con pirfenidona o nintedanib en comparación con un grupo de placebo64–66. En el año 2019, nintedanib fue aprobado por la Administración de Alimentos y Medicamentos de Estados Unidos y las Agencia Europea de Medicinas para el tratamiento de la EPI-ES y para la EPI-FP después de los ensayos SENSCIS67 e INBUILD68. (Tabla 6)69–73.
Terapia antifibrótica
| Fármaco | Mecanismo de acción | ECA | Dosis | Monitoreo | Preferir | Efectos secundarios/toxicidad comunes |
|---|---|---|---|---|---|---|
| Nintedanib | Inhibidor de tirosina quinasaPDGFVEGFFGFR | FPI: TOMORROW, INPULSISEPI-ES: SENSCISEPI-FP: INBUILD | 150 o 100mg dos veces al día | Hemograma y función hepática mensual x 3 meses, luego trimestralmente | Exposición solar prolongada ya sea por lugar de residencia o trabajo | Diarrea, baja de peso, fatiga, elevación de transaminasas |
| Pirfenidona | Antifibrótico por múltiples mecanismosTNF-αTGF-β | FPI: CAPACITY y ASCENDEPI- ES: SLS IIIEPI-AR: TRAIL-170EPIi: μILD71EPI-FP: RELIEF72NH: (NCT02958917)73 | 267mg tres veces al día, titulado a 801mg tres veces al día | Hemograma y función hepática mensual x 3 meses, luego trimestralmente | Uso de anticoagulación.Antecedentes de IAMDiarrea crónica | Intolerancia gastrointestinal, fatiga, baja de peso, elevación de transaminasas, fotosensibilidad, rash cutánea |
Adaptado y modificado de Collins BF y Raghu G. (2019)69.
Se describen los mecanismos de acción principales, ECA (estudios controlados aleatorizados) más importantes, dosis, parámetros a monitorizar, preferencia y efectos adversos de la terapia antifibrótica con nintedanib y pirfenidona.
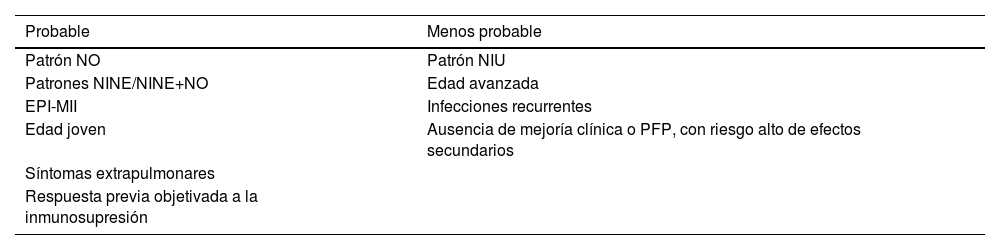
Aunque varios estudios han demostrado un perfil de seguridad adecuado con la combinación de ambas terapias, inmunomoduladora o inmunosupresora y terapia antifibrótica, no hay claridad sobre cuándo combinarlas. Parece adecuado en EPI-ETC con fenotipo FP, pero en NHf no está claro si deben mantenerse ambas terapias o si, ante la progresión bajo terapia inmunomoduladora, debe interpretarse como fracaso terapéutico y cambiar a terapia antifibrótica como en EPI-FP26. En la tabla 7 se describen los beneficios vs. Las complicaciones de mantener la terapia antiinflamatoria en pacientes con EPI-FP.
Beneficio de terapia inmunosupresora en EPI-FP
| Probable | Menos probable |
|---|---|
| Patrón NO | Patrón NIU |
| Patrones NINE/NINE+NO | Edad avanzada |
| EPI-MII | Infecciones recurrentes |
| Edad joven | Ausencia de mejoría clínica o PFP, con riesgo alto de efectos secundarios |
| Síntomas extrapulmonares | |
| Respuesta previa objetivada a la inmunosupresión |
Adaptado y modificado de Rajan SJ et al. (2023)26. Evaluación para decidirentre suspender o mantener la terapia inmunosupresora en pacientes conenfermedad pulmonar intersticial fibrosante progresiva (EPI-FP) dependede factores clínicos y radiológicos específicos.Abreviaturas: NO: neumoníaen organización; NIU: neumonía intersticial usual; NINE: neumonía intersticial no específica; EPI: Enfermedad pulmonar intersticial; MII: miopatíainflamatoria idiopática; PFP: pruebas de función pulmonar.
En resumen, las enfermedades pulmonares intersticiales representan un desafío diagnóstico y terapéutico debido a su variabilidad etiológica y clínica. A lo largo de este análisis, se destaca la importancia de la tomografía computarizada de alta resolución, de la opinión del comité multidisciplinario en el proceso diagnóstico, así como la evolución en la clasificación de estas enfermedades. Es necesario discutir las opciones de tratamiento, con énfasis en la importancia de considerar la etiología, el patrón radiológico, el comportamiento clínico de cada paciente junto a la histopatología para una gestión óptima de estas complejas afecciones respiratorias.
Conflictos de interésEl autor no declara ningún conflicto de interés para esta publicación.