




La Fibrosis Quística (FQ) es una enfermedad hereditaria autosómica recesiva. La detección precoz sumado a medidas de intervención temprana han modificado el curso de esta enfermedad con mejorías en su sobrevida, lo que ha llevado a una población creciente de pacientes de 18 años. La mutación genética determina una alteración en una Proteína Reguladora de Conductancia Transmembrana (CFTR) que afecta a numerosos órganos y sistemas, pero el compromiso pulmonar es el que causa mayor morbimortalidad. El germen más frecuente que infecta a adultos es Pseudomonas aeruginosa y si bien hay una serie de medidas para el manejo de la infección crónica por Pseudomonas las terapia dirigida a la restauración de la función de la proteína CFTRhatomadorelevancia. Cuando la falla respiratoria progresa, la única alternativa disponible es el trasplante pulmonar que mejora la sobrevida y la calidad de vida en estos pacientes.
Cystic Fibrosis (CF) is an autosomal recessive hereditary disease. Early detection combined with early intervention measures have changed the course of this disease with improvements in their survival which has led to a growing population of patients 18 years. The genetic mutation determines an alteration in Transmembrane Conductance Regulator protein (CFTR) that affects many organ systems, but the pulmonary involvement is causing increased morbidity and mortality. The most common pathogen that infects adults is Pseudomonas aeruginosa and while there are a number of measures for the management of chronic Pseudomonas infection of therapy aimed at restoring the function of the CFTR protein has gained importance. When respiratory failure progresses only alternative available is lung transplantation improves survival and quality of life in these patients.
La Fibrosis Quística (FQ) es una enfermedad hereditaria autosómica recesiva, que se presenta predominantemente en raza blanca, con una incidencia que se estima en Chile de 1/8000 recién nacidos vivos. Según datos del MINSAL del año 2014, el sistema público de salud registra 395 pacientes y de ellos 162 son mayores de 18 años (41%)1.
La enfermedad es producida por una mutación en el gen que codifica para la Proteína Reguladora de Conductancia Transmembrana de la Fibrosis Quística (CFTR), ubicado en el brazo largo del cromosoma 7.
Se describen a la fecha más de 2000 mutaciones, siendo la más frecuente la delta F508. Sin suficientes copias funcionales de la proteína CFTR en sus membranas celulares, las células epiteliales no pueden bombear suficiente agua en las secreciones, por lo que éstas son demasiado espesas y viscosas y tienden a obstruir los conductos de diversos órganos, especialmente la vía aérea pequeña en los pulmones.
Esta obstrucción prepara el escenario para la inflamación, la infección secundaria y la eventual destrucción del tejido que lleva finalmente a la producción de bronquiectasias que son características de la FQ y las infecciones que son la causa final de muerte de estos pacientes.
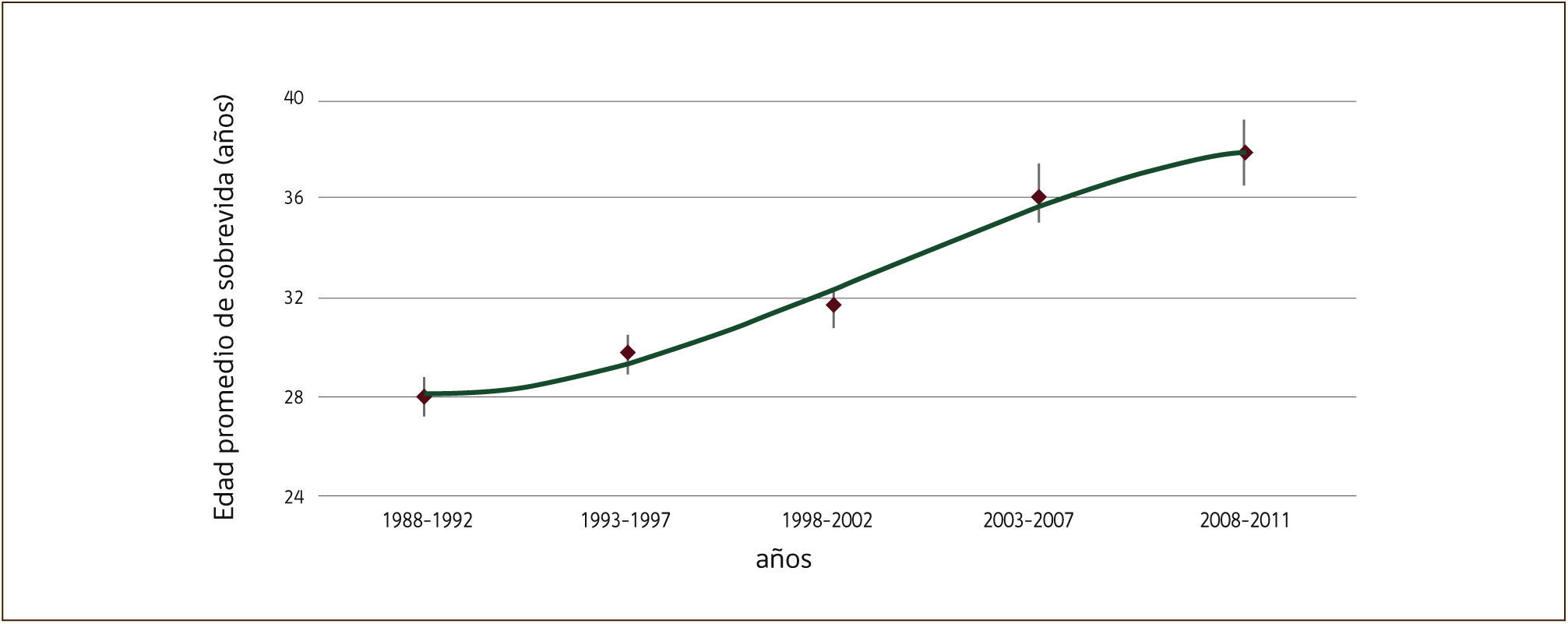
El pronóstico de los pacientes con FQ ha mejorado notablemente en las últimas décadas debido al diagnóstico precoz y a los tratamientos médicos que han modificado el curso de esta enfermedad, tanto previniendo como retardando sus complicaciones. La mediana de supervivencia ha pasado de tan solo un año en 1950 a 37,4 años en 2012, según el Registro de Pacientes de la Fundación Americana para la FQ (Figura 1). El resultado es una población creciente de adultos mayores de 18 años que, en Estados Unidos, ha llegado a representar el 45% de toda la población con FQ1.
GENÉTICA
La FQ es causada por una mutación de un gen localizado en el brazo largo del cromosoma 7 que codifica para una proteína llamada Regulador de Conductancia Transmembrana de la Fibrosis Quística (CFTR)2. Esta, es una proteína que funciona como un canal que actúa principalmente como un transportador de cloro. Expresándose en la membrana apical de los epitelios secretores (pulmón, páncreas, intestino, glándulas sudoríparas, conductos biliares y conductos deferentes). La alteración de este canal determina un aumento del cloro en el intracelular y de una absorción marcada del sodio intraluminal, el que arrastra agua. Lo anterior produce un espesamiento de las secreciones de los epitelios comprometidos, con mal funcionamiento de los cilios y daño en los órganos afectados.
Se describen 6 clases de mutaciones del CFTR:
1- Defectos en la síntesis del CFTR.
2- Defectos en el procesamiento.
3- Defectos en la regulación.
4- Defectos en la conducción.
5- Defecto parcial en la producción o en el procesamiento.
6- Defectos en la regulación de otros canales.
Como el defecto se hereda en forma autosómica recesiva requiere que ambos padres sean portadores del gen defectuoso y la probabilidad de tener un hijo es de un 25% con cada embarazo.
MANIFESTACIONES CLÍNICASLa mutación del CFTR provoca un trastorno en el transporte de sodio, cloro, bicarbonato que favorece secreciones espesas en el pulmón, intestino, páncreas, hígado y aparato reproductor que conducen a su daño3. Se han descrito más de 2000 mutaciones y por lo anterior su expresión fenotípica es variable dependiendo de la o las mutaciones presentes.
-FQ típica o clásica: La mayoría de estos pacientes tienen compromiso de múltiples sistemas (pulmonar, sinusal, pacreático, aparato reproductivo, etc.) y tiene test de sudor positivo.
- FQ no clásica: Se da más frecuentemente en pacientes adultos, que podrían tener test de sudor normal o indeterminado, enfermedad pulmonar leve y con poco compromiso gastrointestinal. Tienen una alta frecuencia de mutaciones CFTR inusuales4.
Manifestaciones no respiratorias:1) Enfermedad pancreática:
- Insuficiencia:
En la FQ los niveles de cloro luminales son insuficientes para el intercambio con bicarbonato, por lo que se produce insuficiente jugo pancreático con tapones y obstrucción de los conductos pancreáticos con activación prematura de las enzimas causando daño y finalmente destrucción del tejido pancreático. Su daño crónico produce fibrosis e insuficiencia pancreática exocrina5.
- Diabetes relacionada con la FQ (CFRD):
La prevalencia de diabetes es 100 veces mayor en pacientes con FQ que en la población general y se incrementa con la edad. Un 20% de los adolescentes y un 40 a 50% de adultos con FQ tiene CFRD6-7. Los pacientes con CFRD tienen peor función pulmonar, malnutrición, se hospitalizan más y tienen mayor mortalidad8.
La recomendación actual es realizar un screening anual con un TTOG desde los 10 años9. Su manejo incluye una ingesta calórica adecuada, ejercicio regular e inicio temprano de insulina.
- Pancreatitis:
Se desarrolla en un 10% de pacientes con suficiencia pancreática y es rara entre pacientes con Insuficiencia pancreática10. Se puede manifestar como pancreatitis aguda, pancreatitis aguda recurrente y pancreatitis crónica. Además, puede ser un signo clínico que oriente al diagnostico de FQ en pacientes adultos o en pacientes con cuadros clínicos leves o atípicos de la enfermedad11.
2) Síndrome de obstrucción intestinal distal (DIOS)
Ocurre hasta en un 15% de adultos con FQ. Es más común en pacientes con genotipos severos y enfermedad pulmonar avanzada. Se caracteriza por episodios recurrentes de obstrucción parcial o completa de intestino por material mucoso y fecal espeso a nivel de la región ileocecal. El paciente relata dolor abdominal en el cuadrante inferior derecho puede haber nauseas, vómitos y distensión abdominal. Al examen puede haber masa palpable en el cuadrante inferior derecho. Un manejo médico precoz puede evitar la cirugía y este incluye hidratación adecuada, desimpactación fecal con polietilenglicol via oral o por sonda nasogástrica1.
3) Enfermedad hepática:
Con la mejoría en la supervivencia las manifestaciones hepáticas de la enfermedad emergen como un problema médico cada vez más importante y constituyen en la actualidad la 3era causa de muerte.
La incidencia de enfermedad hepática es de un 27 a 35%, pero la progresión a cirrosis no es común y ocurre entre un 3 a 7% de los pacientes12, diagnosticándose habitualmente entre los 10-20 años. La cirrosis biliar focal es la manifestación hepática patognomónica como resultado de una obstrucción biliar y fibrosis periportal progresiva. Con el tiempo esta puede progresar a cirrosis multilobular con hipertensión portal clínicamente significativa y sus complicaciones asociadas13.
4) Enfermedad renal:
La incidencia de injuria renal aguda en niños con FQ es 100 veces más frecuente que la población general (0,075 casos por 10.000 niños por año)14. La enfermedad renal crónica en pacientes con fibrosis quística se asocia con aumento de la edad, sexo femenino y mala función pulmonar15.
La hipertensión es rara en pacientes con FQ y se ve más frecuentemente en pacientes con CFRD y aquellos que han recibido un trasplante pulmonar8. El manejo de pacientes con CFRD e HTA es similar a pacientes con DM con la excepción de que la restricción de sal no es necesaria9.
5) Infertilidad:
A pesar de que la espermatogénesis no esta afectada, el 95% de los hombres con FQ es infértil por azoospermia segundaria a agenesia de conductos deferentes e hipoplasia de vesículas seminales, conductos eyaculadores y epidídimo16. Por otro lado, las mujeres con FQ son fértiles o manifiestan discreta diminución de su fertilidad debido a malnutrición y alteración del moco cervical17. En general los resultados son favorables si el VEF1 antes de embarazo es mayor a 50-60% del valor predicho18.
6) Compromiso osteoarticular
Osteopenia y Osteoporosis: La pérdida de la masa ósea en pacientes con FQ parece ser multifactorial (uso de corticoides, hipovitaminosis D, infecciones recurrentes, malnutrición, etc) y puede estar presente hasta en un 75% de los pacientes adultos con FQ19 y por lo anterior, pacientes deberían tener una evaluación a partir de los 10 años1. Estrategias preventivas que se recomiendan: suplementar con vitaminas D, K y calcio; mejorar estado nutricional, buen control glicémico en pacientes con CFRD, tratamiento agresivo de las exacerbaciones pulmonares y ejercicio. Los Bifosfonatos se recomiendan en pacientes con T score o Z score de -2 o menos; o -1 en pacientes con antecedentes de fractura que esperen trasplante pulmonar8.
Artropatía asociada FQ: Presente en un 2-9% de los pacientes y se manifiesta por cuadros de dolor e inflamación de articulaciones20.
7) Cáncer:
Estudios sugieren que pacientes con FQ tienen mayor riesgo de cánceres gastrointestinales que la población general y este aumenta a mayor edad. En un estudio de base de datos del registro de US entre 1990-1999, pacientes con FQ no trasplantados tenían un significativo mayor riesgo de cáncer del tracto gastrointestinal (23.0 v/s 4.5 esperado; SIR 5.1, 95% CI 3.2-7.6)8,21. Entre estos, el mayor riesgo fue de intestino delgado, colon y tracto biliar.
Manifestaciones pulmonaresEn la FQ la mayor viscosidad de las secreciones disminuye el aclaramiento mucociliar y predispone a infección bacteriana generando una respuesta inflamatoria que lleva a obstrucción bronquial de vía aérea, bronquiectasias y falla respiratoria. El grado de compromiso respiratorio depende en parte del genotipo y de factores medioambientales.
Los síntomas respiratorios generalmente aparecen en la infancia pero pueden aparecer en la 2ª o 3ª década de la vida y en mas del 40% de los casos son la sospecha diagnóstica (esto ha cambiado con los métodos de screening neonatales)3. La tos crónica y la expectoración son las manifestaciones principales. Está generalmente aumenta con el tiempo y a medida que progresa el daño pulmonar se vuelve purulenta. Puede aparecer disnea y el paciente presenta exacerbaciones que van aumentando en frecuencia. Los pacientes diagnosticados en la edad adulta pueden tener síntomas más larvados y una función respiratoria normal.
La Rx tórax puede ser normal, pero a medida que progresa el daño pulmonar pueden aparecer hiperinsuflación pulmonar, engrosamiento peribronquial e impactos mucosos. Posteriormente aparecen bronquiectasias de predominio en los lóbulos superiores.
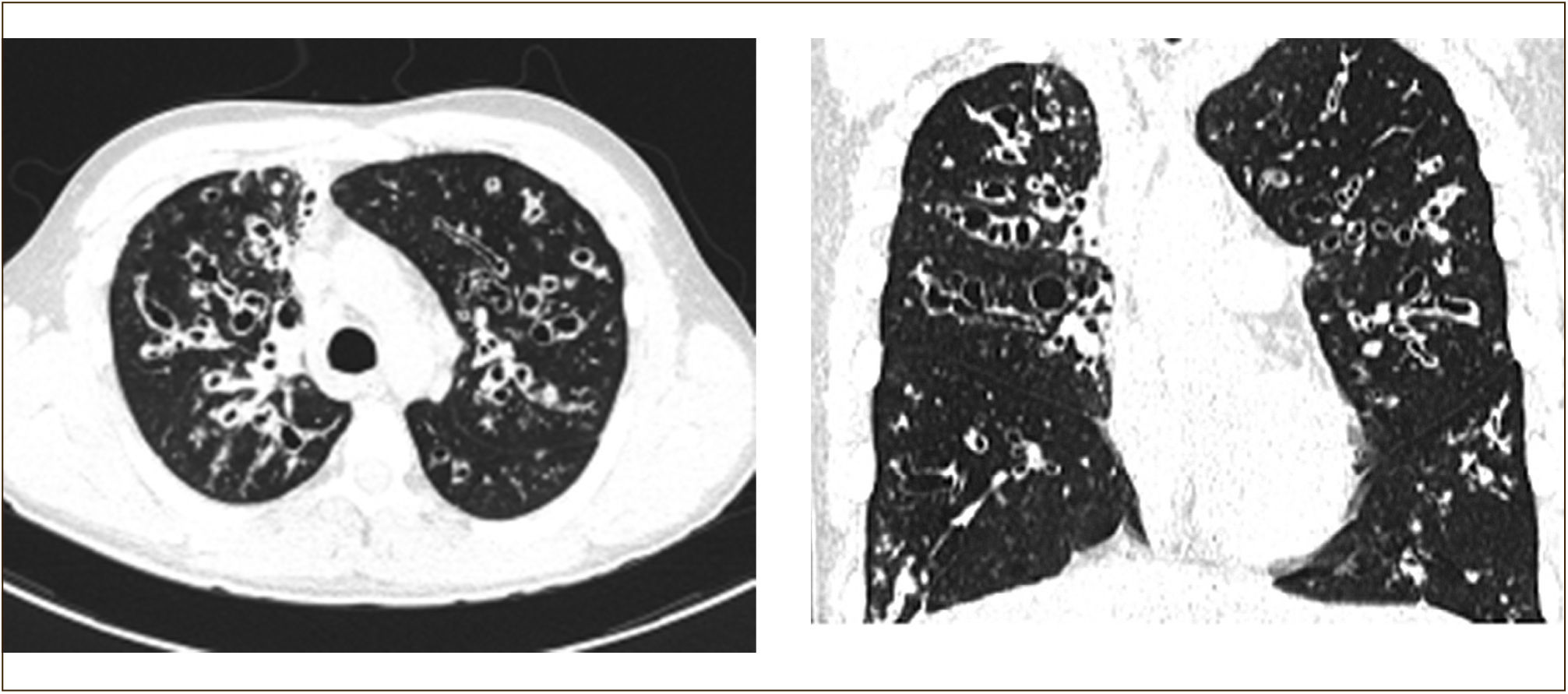
Se recomienda realizar TAC de tórax en para definir extensión de bronquiectasias, y es de utilidad en determinadas complicaciones (neumotórax, infección por micobacterias, entre otras)22 (Figura 2). Otras complicaciones respiratorias no infecciosas presentes en FQ son neumotórax, Aspergilosis Broncopulmonar Alérgica, hemoptisis.
DIAGNÓSTICO
El diagnóstico se basa en la presencia de una característica fenotípica compatible con FQ más una prueba de laboratorio que refleje disfunción del CFTR23.
Características fenotípicas:- -
Cuadro clínico compatible
- -
Historia familiar
- -
Tamizaje neonatal positivo (tripsina inmunoreactiva)
- -
Ausencia bilateral de conductos deferentes
- -
Test de sudor positivo (en 2 ocasiones)
- -
Estudio genético positivo (2 mutaciones del CFTR)
- -
Prueba del potencial nasal diferencial positivo
El test de sudor es el examen fundamental de comprobación diagnóstica. La técnica estándar y confirmatoria es la de Gibson y Cooke, que consiste en recolección del sudor inducida por Iontoforesis con pilocarpina, midiendo el cloro con cloridómetro digital1.
Estudio genético
Si existe duda diagnóstica puede ser un examen confirmatorio si se encuentra mutación en los 2 alelos. La mayoría de los expertos sugiere además hacerlo en todos los casos de FQ confirmada ya que tiene implicancias pronósticas y de tratamiento.
Los paneles de estudio genético convencionales detectan 36 mutaciones más frecuentes según el Cystic Fibrosis Consortium. Pero en pacientes adultos o con cuadros clínicos atípicos, si este screening genético inicial es negativo, se pueden realizar estudios genéticos ampliados para confirmar la enfermedad.
Estudio de la diferencia de potencial nasal
De utilidad en pacientes con FQ no clásica, con test de sudor indeterminado y estudio genético no diagnóstico. Mide la diferencia de potencial eléctrico a nivel de la mucosa nasal. Este método requiere de estandarización y preparación por lo que se realiza solo en centros muy especializados.
Tamizaje: Consiste en la determinación de tripsinógeno inmuno reactivo en el período neonatal. Se están haciendo esfuerzos en incorporar esta técnica en nuestro país.
EXACERBACIONESEl 85% de los pacientes que fallecen por FQ, es debido a una complicación pulmonar, generalmente con infección secundaria. La enfermedad pulmonar comienza tempranamente en la vida con trastorno del clearance mucociliar e infecciones recurrentes, con una declinación progresiva de la función pulmonar con episodios de deterioro de sus síntomas que son definidas como exacerbaciones. Estas tienen impacto negativo en los pacientes tanto en la función respiratorio, en su calidad de vida y un alto impacto económico en el tratamiento.
Los gérmenes más frecuentes que infectan a los pacientes con FQ son: P. aeruginosa, Burkholderia spp., Sthapilococcus aureus y con menos frecuencia otros patógenos gram-negativo como Stenotrophomonas maltophilia, Achromobacter xylosoxidans, Ralstonia, Cupriavidus, y Pandoraea spp y los hongos filamentosos como aspergillus spp32. Nuevos patógenos están aumentando en prevalencia y están asociados con aumento en morbilidad y mortalidad. Tales patógenos incluye: MRSA, Mycobacteria abscessus, Burkholderia dolosa, Burkholderia cepacia complex.
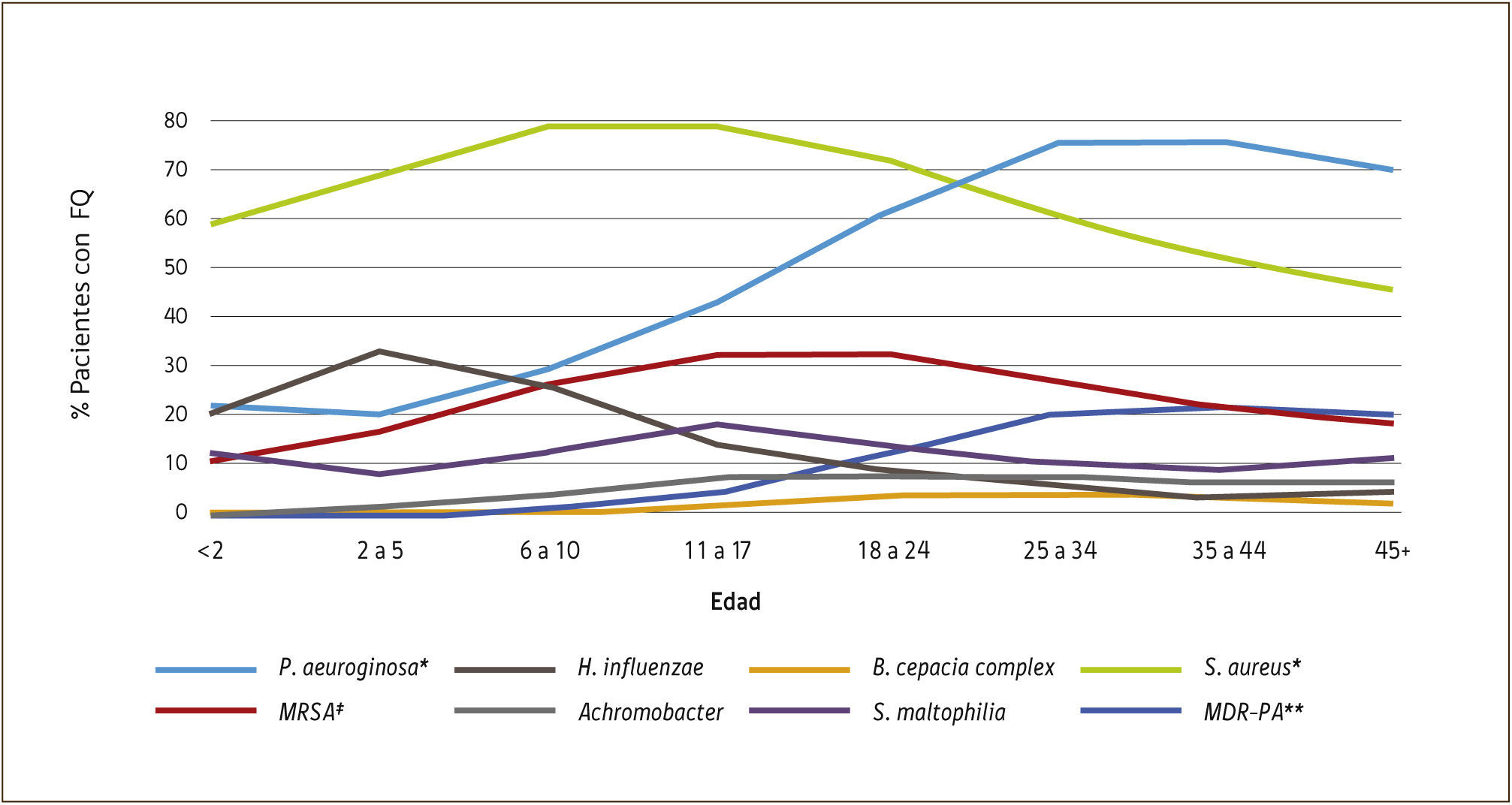
El Registro de pacientes de Fundación para la Fibrosis Quística en EE.UU. provee datos anuales y afirma que el Sthaphilococcus aureus es el patógeno más común en las primeras dos décadas de la vida, mientras que MSSA es más prevalente que el MRSA, la prevalencia de éste último es mayor entre los 11 y los 24 años de edad. La P. Aeruginosa es detectada en más del 20% de jóvenes y cerca del 80% de los adultos son infectados con este patógeno (Figura 3). La prevalencia de organismos gram negativos incluyendo Stenotrophomonas maltophilia, Achromobacter xylosoxidans, B. Cepacia complex, aumenta con la edad24. Los pacientes con FQ también aumenta su riesgo de colonización con hongos filamentosos, incluidos Aspergillus fumigatus, Scedosporium apiospermum y Aspergillus terreus.

Las exacerbaciones se caracterizan por aumento de signos y síntomas pulmonares y de las secreciones de vías aéreas, manifestaciones generales e infiltrados en la radiografía. Hay un cambio en la clínica habitual del paciente que hace necesaria una intervención terapéutica con el fin de revertir los cambios observados en relación con su situación basal previa.
Para el tratamiento de las exacerbaciones existen varias guías y recomendaciones, las guías publicadas en el 2009 por la “Cystic Fibrosis Foundation” recomienda25:
1.- Lugar de tratamiento: Elegir el mejor lugar para el tratamiento antibiótico y drenaje de secreciones. Hoy día las terapias antibióticas endovenosas podrían realizarse en domicilio si existiera en éste todas las medidas necesarias. Ante cualquier duda de lo anterior es necesario hospitalizar.
2.- Terapias de clearance de la vía aérea: Es el segundo gran pilar del tratamiento, después del antibiótico, existiendo una absoluta unanimidad en su aplicación intensiva durante las exacerbaciones, con un mínimo de tres sesiones diarias.
La mayoría de los ensayos han investigado los efectos de diferentes tipos de técnicas de fisioterapia en mejoría a corto plazo en la función de pulmón y producción de esputo:
a) Fisioterapia respiratoria convencional: drenajes bronquiales, la percusión-vibración torácica, la tos eficaz y la espiración forzada.
b) Máscara de presión positiva espiratoria (PPE).
c) Flutter®: instrumento que produce una vibración de la vía aérea y ayuda a desprender el moco y a su movimiento hacia la tráquea.
d) Compresión torácica de alta frecuencia: se aplica con un chaleco conectado a una bomba mecánica que genera un flujo de aire oscilatorio entre 5 y 20Hz. (alto costo).
3.-Antibióticos: El tratamiento antibiótico de una exacerbación en FQ debe estar dirigido al patógeno aislado en el último cultivo realizado considerando su sensibilidad. Si no hay disponible un cultivo reciente, la cobertura antibiótica deberá incluir tratamiento para Staphylococcus y Pseudomonas species. La mayoría de los centros eligen una cefalosporina de tercera generación con efecto antipseudomónico o un carbapenémico más un aminoglicósido. El tratamiento debe ser por al menos 2 a 3 semanas en dosis altas. La P. aeruginosa es uno de los gérmenes frecuentes causal de exacerbación. Si bien hay discusión entre uso de terapia asociada o bien monoterapia para este tratamiento la CFF recomienda uso de terapia bi asociada, debido a que no hay suficiente evidencia para uso de monoterapia26.
Los antibióticos recomendados:
H. influenzae: Amoxicilina-Clavulánico o una cefalosporina de 2ª o 3ª generación.
S. aureus multisensible: Cloxacilina o flucloxacilina, por vía oral en casos leves, o cloxacilina intravenosa en exacerbaciones graves.
P. aeruginosa multisensible: debe ser tratada durante dos semanas con ciprofloxacino por vía oral en exacerbaciones leves o moderadas (20-40mg/kg en niños y 1-2 gr en adultos)8,10; con ceftazidima y aminoglucósidos intravenosos o una penicilina antipseudomonas con un aminoglucósido en casos más graves (recomienda la administración parenteral de 2 antibióticos) durante 14 a 21días. Esta combinación resulta sinérgica y puede evitar la aparición de resistencia. Se necesitan altas dosis de antibiótico, ya que estas drogas no alcanzan buen nivel en el esputo y los pacientes con FQ las metabolizan más rápido.
TERAPIAS EN DESARROLLOSi bien el foco de este artículo está en terapias pulmonares, debemos considerar que la enfermedad es multisistémica y debe plantearse tratamiento específico para cada órgano comprometido.
Desde el descubrimiento del gen CFTR, se han hecho numerosos intentos para revertir el defecto básico producido por la mutación de los genes.
En la actualidad, dos enfoques muy diferentes tienen por objetivo corregir el defecto básico: la terapia génica, dirigida a corregir la alteración genética, y la terapia con moléculas cuyo objetivo es corregir el defecto funcional a nivel de la proteína. La terapia génica está explorando la forma de introducir copias normales del gen en las vías respiratorias de los pacientes con FQ. Consiste en la inserción de un vector recombinante viral al que se le extrae su ADN y se sustituye por el nuevo ADN terapéutico, de manera que este vector viral sirva de vehículo para insertar el ADN en la célula diana24.
Por otro lado, la terapia dirigida a la restauración de la función de la proteína CFTR ha tenido más éxito. En los últimos años se ha comenzado a tener resultados sobre fármacos capaces de actuar directamente sobre la proteína CFTR.
Reparadores de la proteína CFTR:
1.-Ivacaftor (VX-770), ya está disponible como Kalydeco®, mejora el trasporte de iones a través de los canales proteicos, haciendo que el moco secretado sea más fluido, disminuyendo por lo tanto los síntomas respiratorios y el número de infecciones respiratorias graves y está autorizado para tratar la fibrosis quística de pacientes mayores de 6 años, únicamente cuando la enfermedad está causada por la mutación G551D en el gen CFTR33,34.
2.-Lumacaftor (VX-809) se ha administrado como agente único para pacientes adultos con FQ, homocigotos para DF508. Actualmente, lumacaftor está en investigación en combinación con el Ivacaftor, como potenciador que pareciera ser más prometedor, especialmente en mutación DF 50835.
3.-Ataluren (PTC124): reparador de la proteína, aún en fase de investigación, especialmente para mutaciones tipo I.
Rehidratación de superficie de la vía aérea:
1.- Manitol: en polvo seco
2.- Denufusol: activación de un canal de sodio alternativo36
Agentes antiinflamatorios
a) El ibuprofeno se ha demostrado que retrasa la progresión de la enfermedad pulmonar en los niños, pero las preocupaciones sobre los efectos secundarios han limitado su adopción generalizada.
b) La azitromicina, que funciona como un agente anti-inflamatorio e inmunomodulador, ha demostrado eficacia en pacientes con y sin infección crónica P. Aeruginosa.
Tratamientos anti-infecciosos inhalados:
- Tobramicina en polvo seco (TIP), eficaz tanto como la nebulizada para mejorar función pulmonar y disminuir densidad en esputo de P. Aeruginosa
- Polvo seco de Colistin en pacientes colonizados por P. Aeruginosa.
- Aztreonam nebulizado en P. Aeruginosa.
TRASPLANTE PULMONARSi bien la FQ es una enfermedad que afecta la función de múltiples órganos, la falla respiratoria es la causa más frecuente de muertes en estos pacientes27. Según el registro de la Sociedad Internacional de Trasplante de Corazón y Pulmón 2013 la supervivencia promedio de pacientes con FQ que se trasplantan de pulmón es de 8,3 años28.
Cuando referir a programa de trasplante:29 (Tabla 1)
INDICACIONES DE DERIVACIÓN Y TRASPLANTE EN PACIENTES CON FQ
| Criterios de derivación | Criterios de trasplante |
|---|---|
| • VEF1 < 30% o rápido deterioro | • Oxígeno dependiente |
| • Exacerbación que requiere UCI | • Hipercapnia |
| • Aumento frecuencia exacerbaciones que requieren antibióticos | • Hipertensión pulmonar (HTP) |
| • Neumotórax refractario y/o recurrente | |
| • Hemoptisis recurrente no controlada con embolización |
Referencia 29.
1) Un VEF1 < 30% del predicho o deterioro rápido del VEF1 especialmente en mujeres jóvenes a pesar de tratamiento médico óptimo.
2) Exacerbación pulmonar que requiere manejo en unidad de cuidados intensivos.
3) Incremento en la frecuencia de exacerbaciones que requiere terapia antibiótica.
4) Neumotórax recurrente y/o refractario.
5) Hemoptisis recurrente no controlada con embolización.
Cuando trasplantar un paciente con FQ:29 (Tabla 1)
1) Insuficiencia respiratoria
2) Hipercapnia
3) Hipertensión pulmonar
La colonización por Pseudomonas Auereginosa Multi o Panresistente y otros gérmenes como SAMR, gram negativos multi o panresistentes como Stenotrophomonas maltophilia o Achromobacter species (xylosoxidans) no se ha demostrado que tengan peores resultados post trasplante y por lo tanto no son contraindicación para trasplante30.
En contraste, reportes de pacientes infectados por Burkholderia cepacia complex (Bcc) especialmente aquellos colonizados con B cenocepacia tiene mayor mortalidad y complicaciones post trasplante. Lo anterior hace que muchos centros a nivel mundial consideren como contraindicación absoluta la colonización por este germen27.
La colonización por Aspergillus es común en pacientes con FQ y no es considerada una contraindicación para trasplante. Pacientes con Aspergillus pretrasplante y aquellos con ABPA sin cultivo de esputo positivo deben quedar con antifúngico profiláctico post trasplante27
Las causas de muerte en el primer mes post trasplante son por complicaciones quirúrgicas, disfunción primaria de injerto e infecciones mientras que después del primer año el síndrome de bronquiolitis obliterante y las infecciones no asociadas a CMV dan cuenta del 67% de las muertes31.
Un manejo multidisciplinario en el pre y post trasplante que incluya psicoterapeuta, rehabilitación, kinesiólogo, nutriólogo, broncopulmonar, infectólogo son esenciales para el éxito del trasplante.
TRANSICIÓNEl mayor conocimiento de la enfermedad ha logrado que con los años el diagnóstico sea cada vez más temprano, lo que ha permitido el inicio de medidas de intervención precoz que han logrado mejorías significativas en la sobrevida de estos pacientes. El resultado es una población creciente de pacientes con FQ mayores de 18 años.
Entonces se hace mandatorio que los centros pediátricos que atienden pacientes con FQ cuenten con un protocolo que incluya una transición a un centro de adultos, este debe ser un proceso planificado y coordinado. Lo recomendado es que esta transición:
1) Sea de inicio temprano
2) Un proceso flexible
3) Que incluya una estrecha cooperación entre los centros pediátricos y el de adulto que permita evaluar diferencias en los protocolos de tratamiento y procedimientos diagnósticos.
4) Transferencia definitiva entre los 18-20 años de edad.
El proceso de transición permitirá una preparación del adolescente, su familia, del equipo médico de adultos y pediatras que permitan al paciente un mayor grado de independencia, visitas incluyan conocer el centro de adultos y su nuevo equipo médico; visitas del equipo médico adulto al centro pediátrico, etc. Los autores declaran no tener conflictos de interés, en relación a este artículo.