




La sobrevida y la calidad de vida de los pacientes de Fibrosis Quística (FQ) ha mejorado notablemente en las últimas décadas, al punto que ha pasado de ser una enfermedad letal en la infancia, a ser más bien una enfermedad crónica, con la mayoría de los casos en edad adulta. Esto se debe al mejor conocimiento de la patología, a su diagnóstico cada vez más precoz y al manejo preventivo y agresivo de las complicaciones respiratorias y nutricionales, en centros de atención multidisciplinaria especializados. En esta revisión se describe su manejo actual y complicaciones.
The median survival and the quality of life of Cystic Fibrosis patients has increased remarkably in the last decades, changing from a lethal disease to a chronic disease, with a majority of patients in adult age, due to better knowledge of the pathology, precocious diagnosis, and preventive and aggressive management of the respiratory and nutritional complications, realised in specialized and multidisciplinary centers. This article describes the present management of the disease and its complications.
Desde su descripción en 1938 por la Dra. Dorothy Andersen 1, la Fibrosis Quística (FQ) se ha definido como la enfermedad genética letal más frecuente en caucásicos. Sin embargo, en la actualidad, en que más de la mitad de los pacientes de FQ son adultos, parece más adecuado quitarle la connotación de letal y definirla como una enfermedad genética crónica, de manifestación multisistémica y cuyo pronóstico depende de la precocidad del diagnóstico y del manejo terapéutico riguroso en centros especializados.
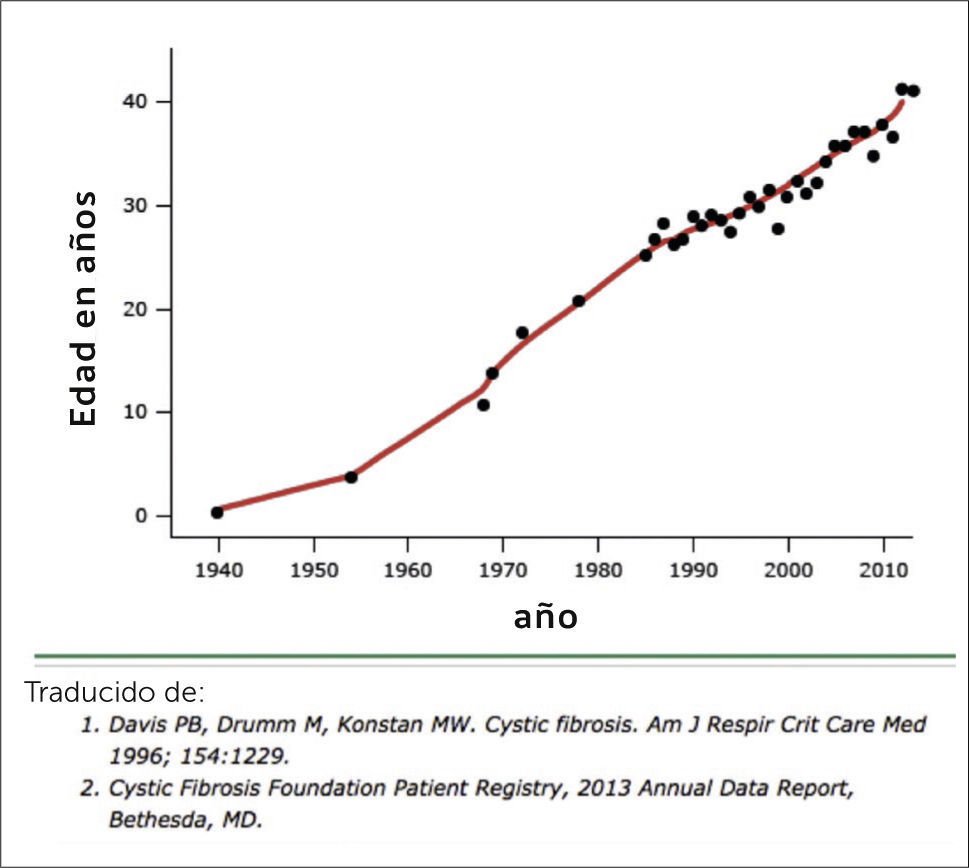
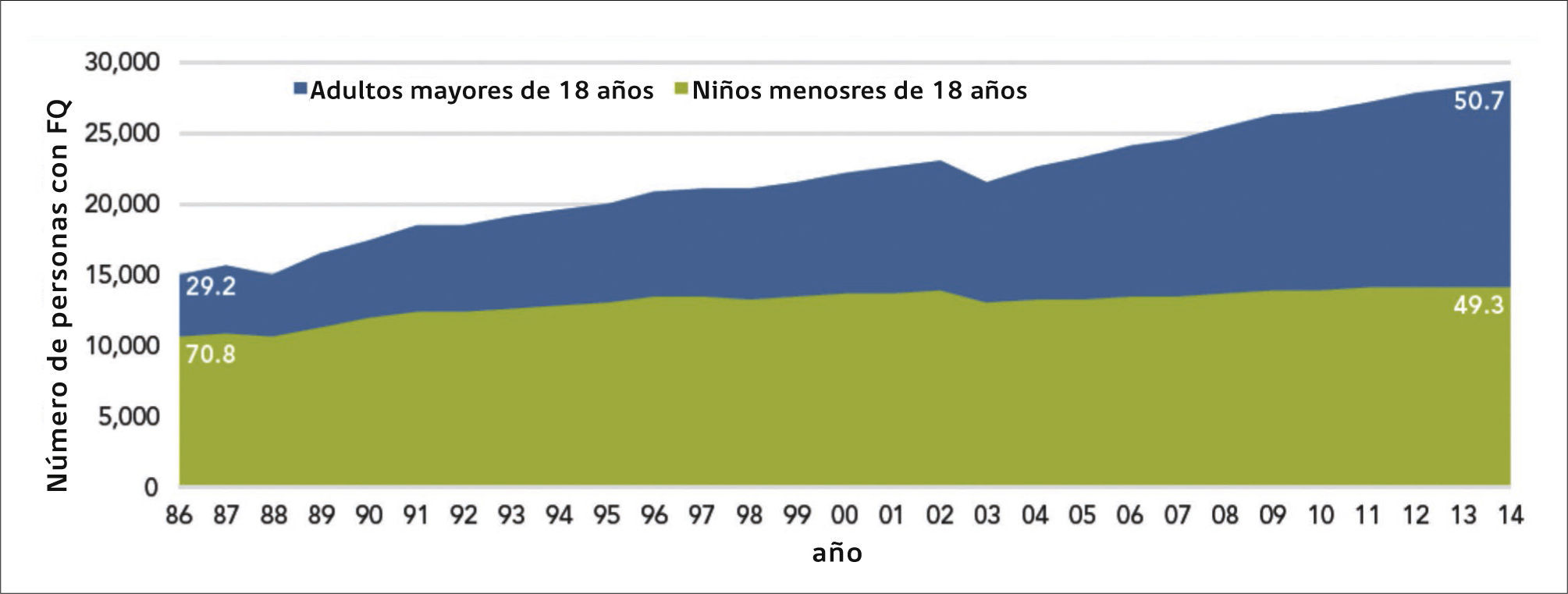
La mediana de sobrevida de los pacientes de FQ ha aumentado considerablemente, llegando en la actualidad a los 41 años (Figura 1), con más de la mitad de los pacientes alcanzando la edad adulta 2 (Figura 2). Este gran avance en el pronóstico vital se debe fundamentalmente al mejor conocimiento de la etiopatogenia y la fisiopatología de la enfermedad, a terapias avaladas en la evidencia de estudios clínicos bien desarrollados, y al manejo médico en centros de FQ con enfoque multisistémico.

 1986-2014 Tomado del Reporte anual 2014 de la Fundación de Fibrosis Quística de Estados Unidos.")
En los últimos años han aparecido nuevas terapias, dirigidas a corregir el defecto básico de la enfermedad, lo que ha creado la gran esperanza de conseguir pronto la cura definitiva de la enfermedad. Mientras ello no ocurra, es fundamental aplicar las terapias dirigidas a prevenir y corregir las complicaciones de la FQ.
La presente revisión tiene como objetivo principal la puesta al día en el manejo terapéutico actual de la enfermedad.
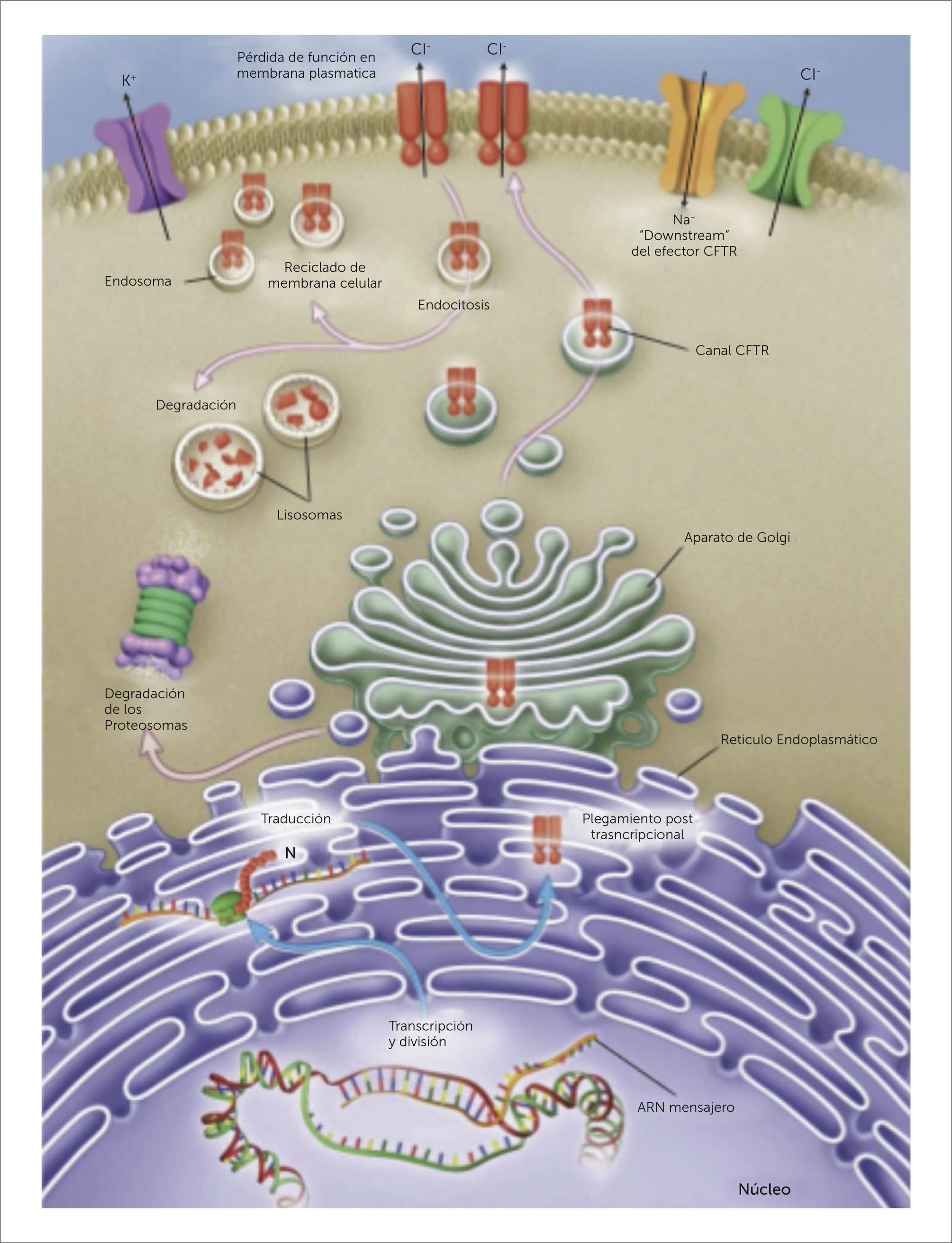
GENÉTICA Y FISIOPATOLOGÍAPara entender las terapias dirigidas a mejorar la sobrevida y la calidad de vida de la FQ, debemos recordar su etiopatogenia y el origen de sus complicaciones. Se sabe que es una enfermedad genética, autosómica recesiva, resultado de la mutación de un gen ubicado en el brazo largo del cromosoma 7, que codifica la producción de una proteína compleja llamada CFTR por su sigla en inglés (“cystic fibrosis transmembrane regulator”), presente en numerosos epitelios y que funciona como un canal o poro para el paso del ión cloro 3,4,7 (Figura 3).

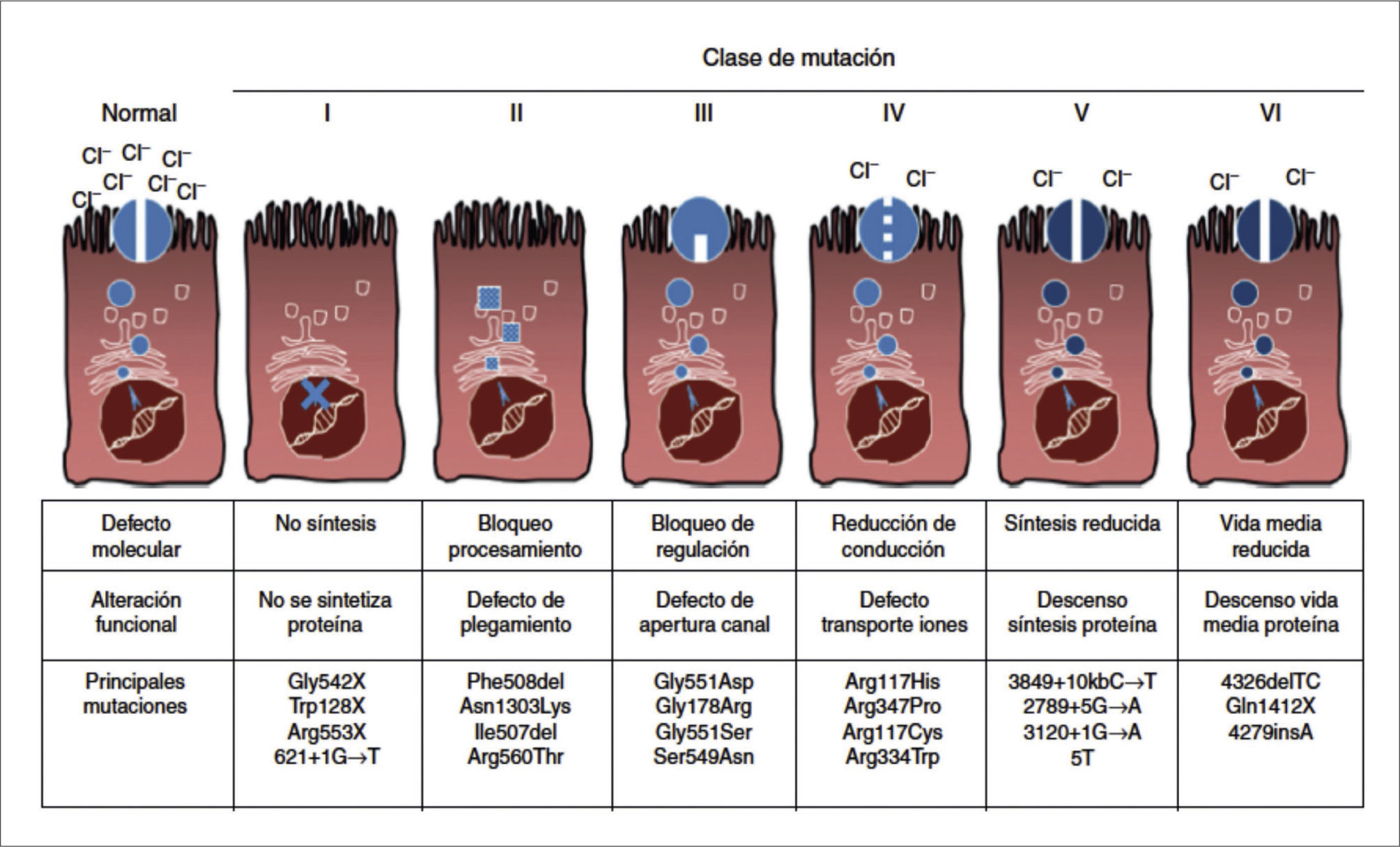
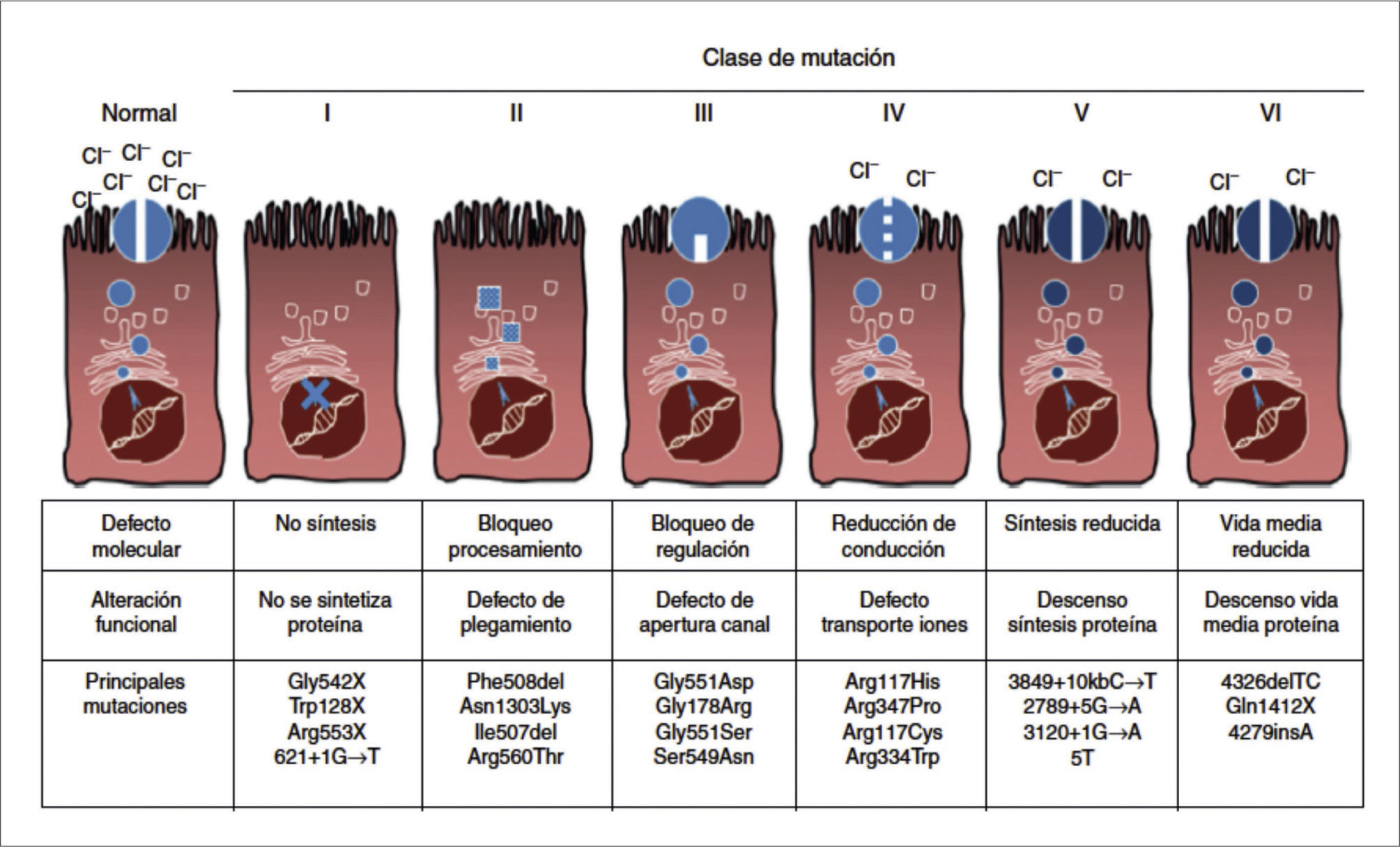
Se han descrito desde su descubrimiento en 1989, 5,6 más de 2000 mutaciones del gen CFTR, que se clasifican en 6 grupos, dependiendo del mecanismo de producción del defecto de la proteína 9 (Figura 4). Las clases I, II y III ocasionan ausencia total del canal del cloro, en cambio las clases IV, V y VI resultan en una falla parcial de la proteína. Si ambas mutaciones de un caso particular son de las tres primeras, el fenotipo resultante será severo, con insuficiencia pancreática. En cambio, la presencia de una mutación de las últimas 3 clases en un alelo, basta para que el fenotipo resultante sea leve, definido por la presencia de suficiencia pancreática 8.
La incidencia reportada es de 1/3500 recién nacidos vivos en caucásicos y de 1/8000 en hispanos.
La falla del CFTR en distintos epitelios explica las manifestaciones multisistémicas, incluyendo las respiratorias, digestivas, hepáticas, metabólicas y reproductivas.
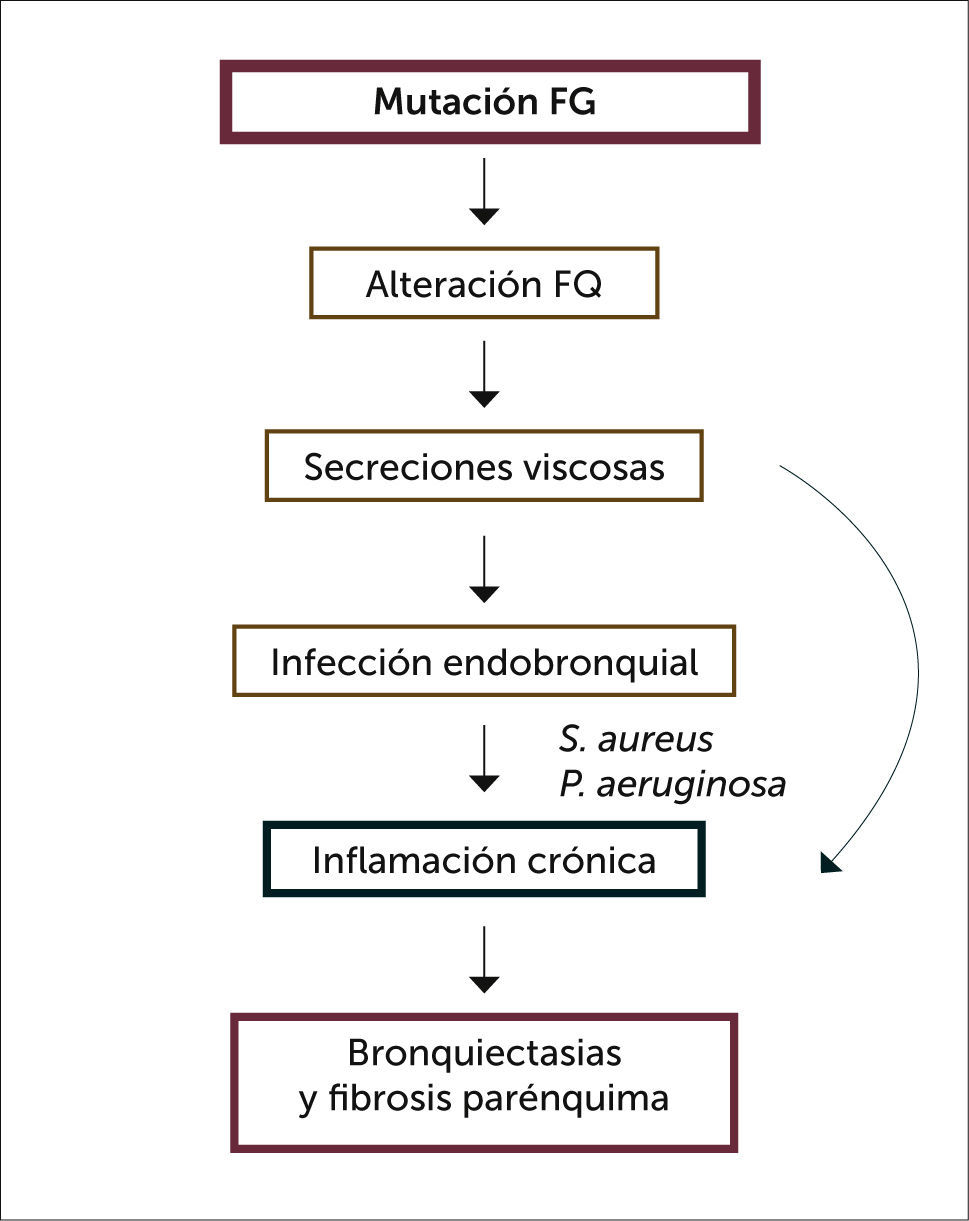
En el epitelio respiratorio, la falla del canal impide la secreción del ión cloro al lumen de la vía aérea, generando la no retención del ión sodio y por ende de agua, produciendo pérdida de hidratación de las secreciones bronquiales 10. Esta alteración de las características del mucus bronquial predispone a la infección por bacterias, especialmente Staphylococcus aureus y Pseudomonas aeruginosa, la que sumada a la respuesta inflamatoria neutrofílica del huésped, terminan produciendo secreciones espesas y viscosas, que obstruyen la vía aérea y destrucción con fibrosis de las paredes bronquiales (Figura 5).
")
Este fenómeno inflamatorio crónico de la vía aérea, causante del daño anatómico y funcional de la FQ, está presente desde las primeras semanas de vida, incluso en ausencia de infección bronquial demostrable en los cultivos de secreción bronquial. Este hecho remarca la importancia de iniciar las terapias para mantener la salud pulmonar lo más precozmente posible 11.
PILARES DEL TRATAMIENTO DE LA FQEl manejo actual de la FQ se sustenta en 5 pilares fundamentales, que no pueden dejar de estar presentes:
- I)
Diagnóstico lo más precoz posible
- II)
Mantener la vía aérea libre de secreción
- III)
Mantener la vía aérea libre de infección
- IV)
Mantener un estado nutricional óptimo
- V)
Manejo en centro especializado multisistémico
I) DIAGNÓSTICO PRECOZ
El ideal es llegar al diagnóstico en los primeros meses de vida, por lo que el pediatra debe estar atento a los síntomas de sospecha (Tabla 1). Sin embargo, el daño pulmonar se inicia ya desde el nacimiento y es muy probable que esperar la aparición de los síntomas nos haga llegar tarde al diagnóstico 11.
INDICACIONES DEL TESUDORST
| -Síntomas respiratorios recurrentes (Neumonías o Sibilancias recurrentes, tos persistente) |
| -Diarrea crónica, mala absorción |
| -Retardo desarrollo pondoestatural |
| -Deshidratación hiponatrémica, hipoclorémica e hipokalémica con alcalosis metabólica |
| -Edema e hipoproteinemia |
| -Sabor salado de la piel |
| -Ileo meconial |
| -Hepatomegalia |
| -Prolapso rectal |
| -Pólipos nasales |
| -Ictericia prolongada del recién nacido |
| -Hermano con FQ |
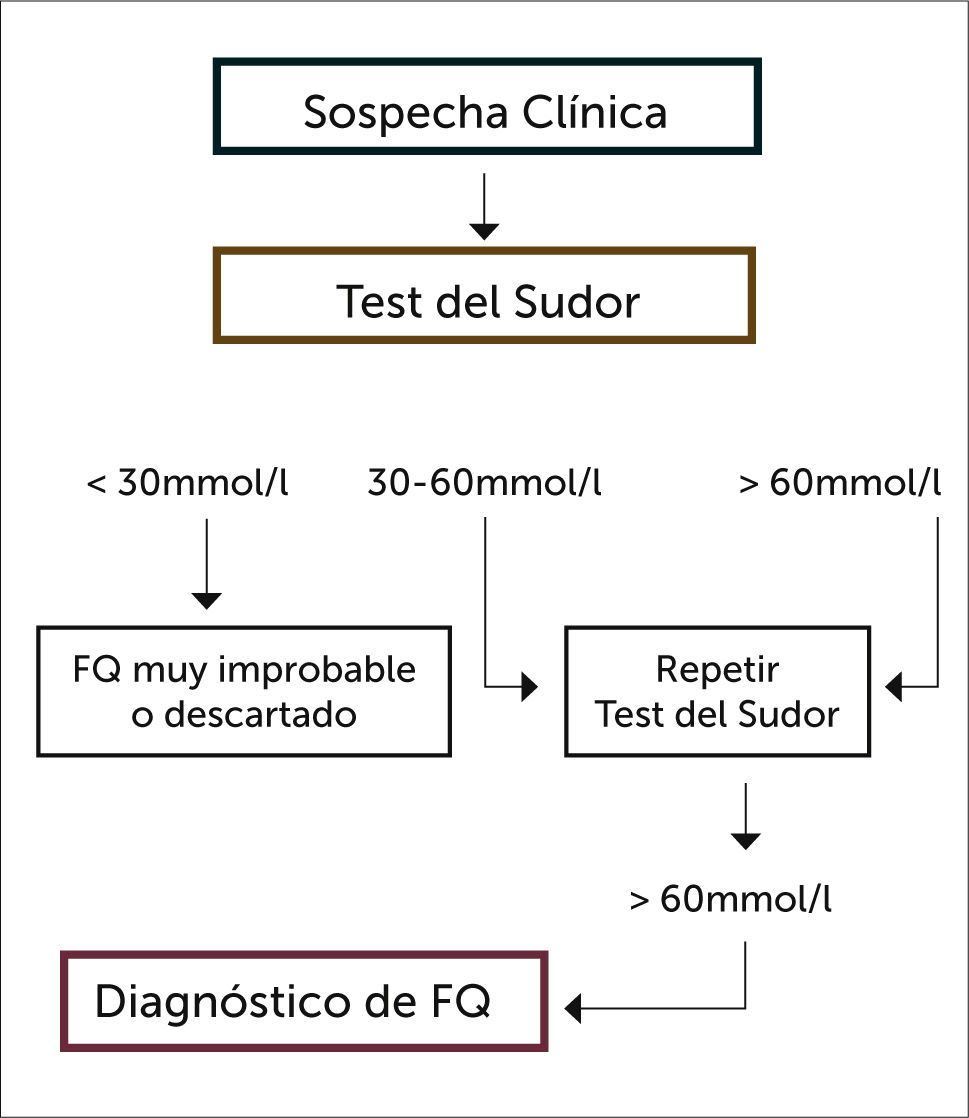
De ahí la importancia del tamizaje neonatal para FQ, que se realiza en la mayor parte de los países desarrollados y que permite detectar todos los casos y por lo tanto la verdadera incidencia de la enfermedad. Las estrategias son variadas, y consisten en tomar Tripsina inmunoreactiva (IRT) de una muestra de sangre del talón al recién nacido de término a las 48 a 72 horas de vida (a los 7 días en el prematuro). Si resulta elevado, (sobre 60 ng/ml) el paso siguiente es repetir IRT a la segunda o tercera semana de vida (nueva muestra), o realizar estudio de mutaciones (de la primera muestra) o medir PAP (proteína asociada a pancreatitis) 12,13. Finalmente, todos los casos sospechosos deben confirmarse con 2 test del sudor con la técnica de Gibson y Cook (Figura 6).

II) MANTENER LA VÍA AÉREA LIBRE DE SECRECIÓN
a) Kinesiterapia respiratoria
Es clave y central en el manejo respiratorio de la FQ. Debe iniciarse desde el momento del diagnóstico y formar parte de la vida diaria del paciente. Se realiza un mínimo de 2 sesiones diarias, al levantarse y al acostarse, aumentando la frecuencia según la necesidad. Los padres deben aprender a realizar las maniobras básicas desde el diagnóstico y luego es el mismo paciente el que las llevará a cabo.
Las técnicas varían y dependen de la edad: en el menor de 3 años, maniobras pasivas de percusión del tórax, bloqueos, vibraciones y drenaje postural. Después de los 3 años, se pueden agregar técnicas de espiración forzada, induciendo al niño a espirar lenta y progresivamente, para llevar las secreciones hacia la vía aérea central. En el niño mayor de 6 años, se le enseñan maniobras de “drenaje autogénico” 14.
b) Agentes farmacológicos que promueven la limpieza de las secreciones
- DNasa I (Dornase alfa): Es una endonucleasa, que rompe las cadenas de DNA liberados por los neutrófilos, responsable de la alta viscocidad de la secreciones en la FQ. Se ha demostrado que mejora la función pulmonar, elevando el VEF1 en un 6% en pacientes FQ mayores de 6 años, y disminuye la frecuencia de las exacerbaciones pulmonares 15.
Estudios de su efecto en niños menores y lactantes, muestran efecto beneficioso, al disminuir el atrapamiento aéreo periférico, demostrando actuar en la vía aérea fina.
La dosis usual es 2.5mg a nebulizar cada 24 horas, diariamente y a permanencia, previo uso de broncodilatadores en aerosol (Salbutamol) para prevenir broncoconstricción.
La mayoría de las guías clínicas lo indican en el paciente sintomático respiratorio mayor de 6 años, con daño pulmonar moderado o severo 16. Esto deriva de que la mayoría de los estudios prospectivos se han realizado en niños mayores, capaces de realizar estudios funcionales (Espirometría). Nuestro centro comparte la idea de que en preescolares y lactantes debemos iniciar el uso crónico de Dornase alfa desde el inicio de los síntomas respiratorios 28–30.
- Solución salina hipertónica al 7%: Se ha estudiado superar la falla de hidratación de la capa acuosa del mucus bronquial, mediante la nebulización de solución hipertónica de NaCl al 7%. Si bien no mejora con sifnificación estadística la función pulmonar, sí demuestra una tendencia sobre el placebo, y se ha demostrado beneficioso, disminuyendo la frecuencia de exacerbaciones. Se usa nebulizando 4ml al 7%, 2 veces al día, previo uso de broncodilatador, de manera permanente. Nuestra indicación, al igual que Dornase alfa, es en el paciente sintomático respiratorio, sin considerar la edad 28–30.
III. MANTENER LA VÍA AÉREA LIBRE DE INFECCIÓN
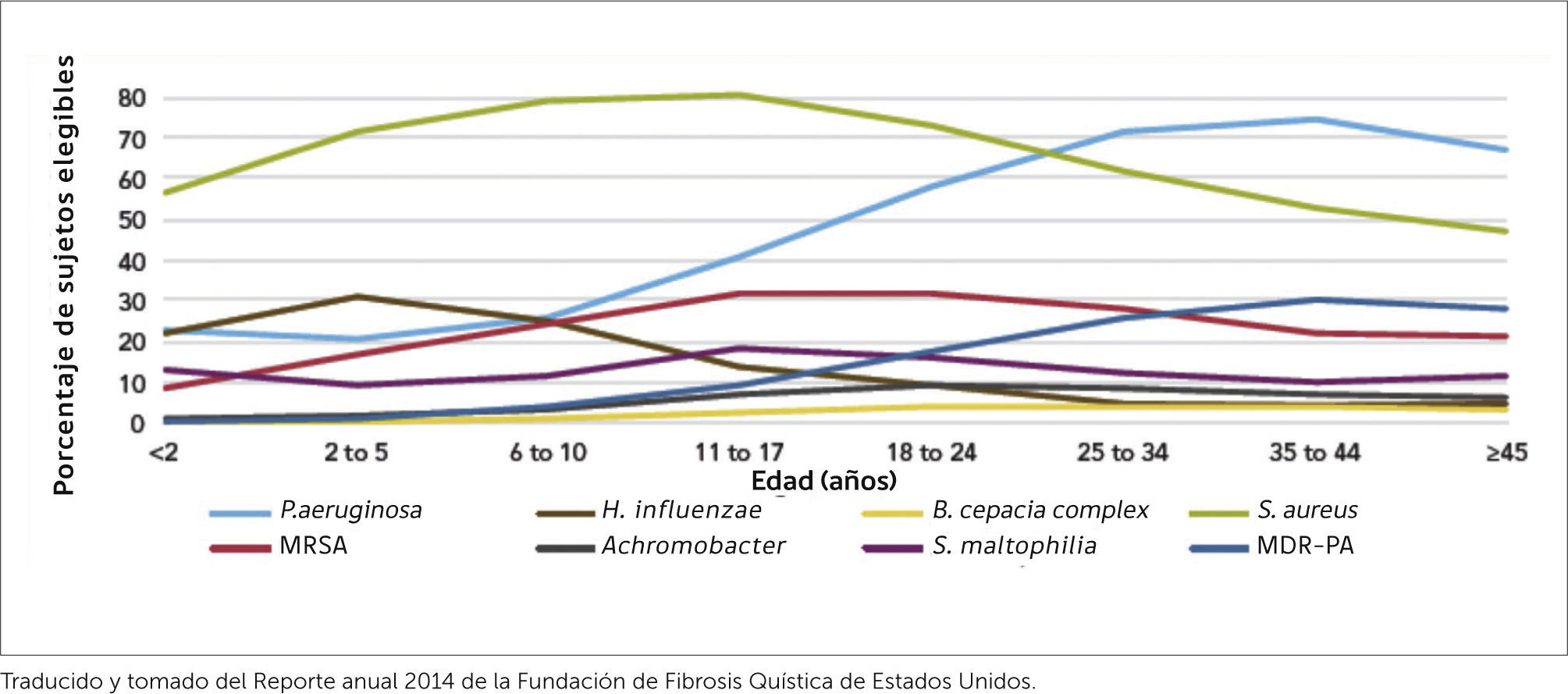
La infección bacteriana endobronquial crónica es el principal factor causante del deterioro progresivo de la función pulmonar y del mal pronóstico de la enfermedad. Pseudomonas aeruginosa es la bacteria más relacionada con dicho deterioro y por lo tanto es clave en el manejo de la FQ, evitar el desarrollo de infección crónica por esta bacteria 20. Debemos recordar que la vía aérea del paciente con FQ se va colonizando con distintos gérmenes, inicialmente con Staphylococcus aureus como el más frecuente y posteriormente Pseudomonas aeruginosaFigura 7.

Inicialmente la infección por Pseudomonas aeruginosa es por bacterias libres, (planctónica), las que van multiplicándose adosadas al epitelio de la vía aérea, a través de Pili o fimbrias de adhesión. En esta etapa inicial, la infección es erradicable con los esquemas antibióticos adecuados. Sin embargo, si la infección no es tratada, con el paso del tiempo (probablemente alrededor de 1 año), se alcanza un número tal de gérmenes (“quorum sensing”), que adquieren la capacidad de liberar al medio externo un biofilm de alginato, que cubre completamente la colonia y que les otorga un verdadero escudo protector, impenetrable para anticuerpos, neutrófilos y antibióticos 21,22. En esta etapa, la infección se hace inerradicable y las terapias antibióticas sólo logran disminuir la cantidad de colonias.
Es importante tener presente que cada vez que una nueva infección endobronquial produce síntomas, lo que llamamos exacerbación aguda, se produce una caída en la función pulmonar, que el tratamiento antibiótico revierte sólo parcialmente y no logra volver a su nivel inicial 26. Así, con cada exacerbación se va reduciendo progresivamente la función pulmonar. Se considera exacerbación aguda, a la presencia de cualquiera de los síntomas y signos siguientes 27:
- •
Nueva tos o aumento de la tos previa
- •
Aumento de expectoración o cambio en su aspecto
- •
Aumento de la signología pulmonar a la auscultación
- •
Disminución del VEF1 en la espirometría
- •
Disminución de apetito
- •
Falta de progreso ponderal
- •
Disnea con ejercicio
- •
Polipnea, retracción torácica, dificultad respiratoria
- •
Fiebre
Para evitar las exacerbaciones y las infecciones crónicas, que van minando la salud respiratoria y la función pulmonar, es fundamental mantener una vigilancia microbiológica de la vía aérea, mediante la toma periódica de cultivos de secreción bronquial. Se recomienda hacer cultivos mensuales en las formas severas de FQ, y cada 3 meses en las formas leves. El laboratorio de microbiología debe tener protocolizado el estudio de muestras de expectoración de pacientes de FQ, para la búsqueda de los principales agentes, como Staphylococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa, Bulkholderia cepacia, Stenotrophomonas maltophilia, Aspergillus fumigatus y Mycobacterias no TBC.
El manejo de la infección endobronquial debe considerar, en consecuencia:
- •
Adelantarse a la aparición de síntomas, con cultivos de expectoración periódicos.
- •
Si el paciente no tiene expectoración, se realiza cultivo faríngeo, que se correlaciona estrechamente con el de vía aérea inferior.
- •
Los cultivos positivos deben ser evaluados a la brevedad para decidir uso de antibióticos.
- •
Mantener registro de todos los cultivos en el tiempo, para calificar cronicidad de la infección.
- •
Definimos como infección crónica el cultivo positivo de un gérmen en más de la mitad de los exámenes en el lapso de 1 año.
- •
Consideramos primoinfección por Pseudomonas aeruginosa, al primer cultivo positivo en un paciente virgen de infección previa, o al primer cultivo positivo después de 1 año libre de infección post terapia de erradicación.
- •
Se ha demostrado que la evaluación de la sensibilidad antibiótica in vitro no se correlaciona con las respuesta clínica en FQ, por lo no se toma en cuenta en la decisión terapéutica.
- •
Los antibióticos inhalados tienen la ventaja de aportar altas concentraciones del fármaco directamente en la vía aérea, sin toxicidad sistémica.
La terapia antibiótica en FQ dependerá entonces de las siguientes circunstancias:
1. Terapia de erradicación de la primoinfección porPseudomonas aeruginosa
Contamos con 3 regímenes muy efectivos y que no requieren de hospitalizar al paciente:
- a)
Colistina, solución para inhalar, 1 millón de unidades 2 veces al día, por 28 días, más Ciprofloxacino oral 20mg/kg /día en 2 dosis por 14 días, o
- b)
Tobramicina, solución para inhalar, 300mg 2 veces al día por 28 días.
- c)
Aztreonam lisina, solución para inhalar, 75mg 3 veces al día por 28 días.
Con estos esquemas terapéuticos inhalados se logra la erradicación de la infección por Pseudomonas aeruginosa en más del 80% de los casos. En aquellos pocos casos en que el cultivo persiste positivo, se realiza una segunda cura antibiótica inhalada, por 3 meses consecutivos 23,25.
2. Terapia de la exacerbación aguda
El esquema terapéutico dependerá del gérmen involucrado y de la intensidad de la exacerbación. Las exacerbaciones por Pseudomonas aeruginosa deben tratarse siempre por vía endovenosa, sean éstas leves o severas, con 2 medicamentos, habitualmente un betalactámico y un aminoglicósido. Las exacerbaciones por otros gérmenes dependerá de la intensidad de los síntomas la decisión terapéutica: en las exacerbaciones leves, sin dificultad respiratoria y sin compromiso del estado general, se pueden utilizar esquemas orales, en las severas siempre endovenosos.
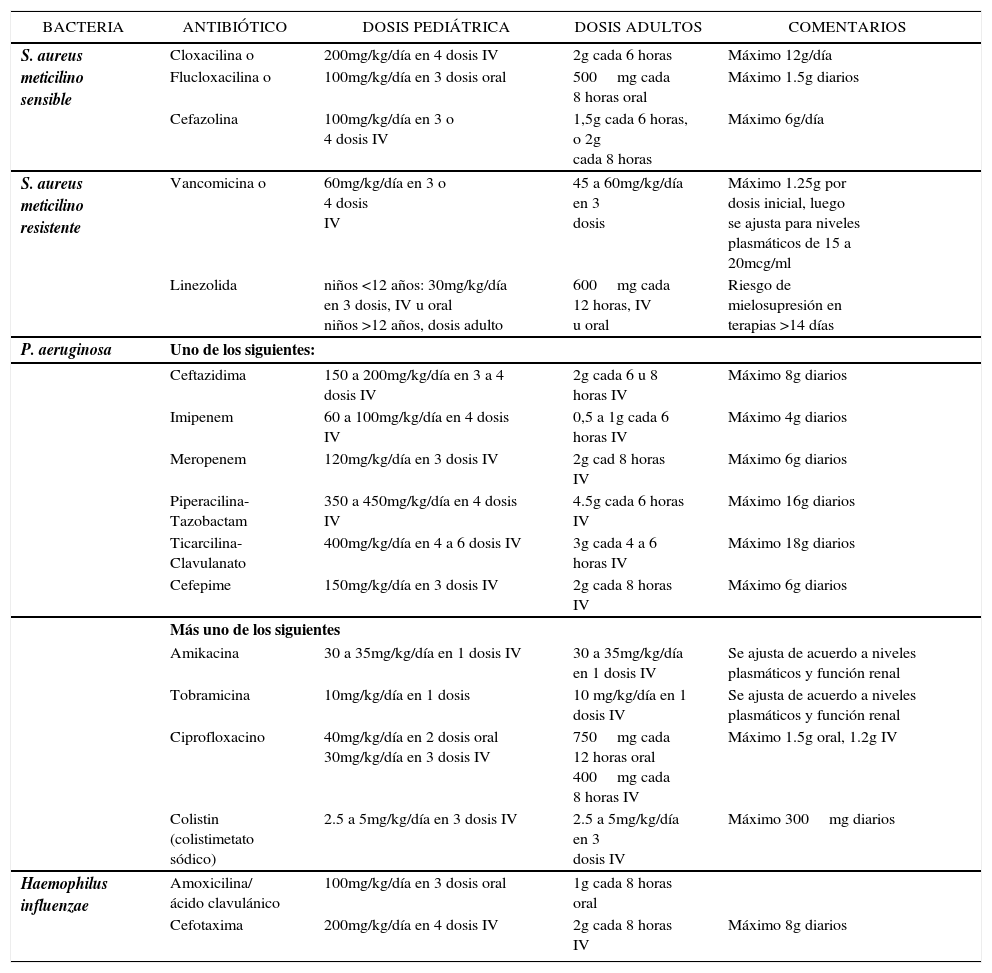
No hay consenso en cuánto a la duración del tratamiento de exacerbación aguda, la evidencia demuestra que no hay mayor cambio clínico ni de la función pulmonar después de 10 días de terapia endovenosa, por lo que esquemas prolongados no debieran usarse. Pensamos que 10 a 14 días es un tiempo suficiente y prudente 28,29. Los esquemas para los gérmenes principales aparecen en la Tabla 2.
ANTIBIÓTICOS PARA EL TRATAMIENTO DE LA EXACERBACIÓN AGUDA
| BACTERIA | ANTIBIÓTICO | DOSIS PEDIÁTRICA | DOSIS ADULTOS | COMENTARIOS |
|---|---|---|---|---|
| S. aureus meticilino sensible | Cloxacilina o | 200mg/kg/día en 4 dosis IV | 2g cada 6 horas | Máximo 12g/día |
| Flucloxacilina o | 100mg/kg/día en 3 dosis oral | 500mg cada 8 horas oral | Máximo 1.5g diarios | |
| Cefazolina | 100mg/kg/día en 3 o 4 dosis IV | 1,5g cada 6 horas, o 2g cada 8 horas | Máximo 6g/día | |
| S. aureus meticilino resistente | Vancomicina o | 60mg/kg/día en 3 o 4 dosis IV | 45 a 60mg/kg/día en 3 dosis | Máximo 1.25g por dosis inicial, luego se ajusta para niveles plasmáticos de 15 a 20mcg/ml |
| Linezolida | niños <12 años: 30mg/kg/día en 3 dosis, IV u oral niños >12 años, dosis adulto | 600mg cada 12 horas, IV u oral | Riesgo de mielosupresión en terapias >14 días | |
| P. aeruginosa | Uno de los siguientes: | |||
| Ceftazidima | 150 a 200mg/kg/día en 3 a 4 dosis IV | 2g cada 6 u 8 horas IV | Máximo 8g diarios | |
| Imipenem | 60 a 100mg/kg/día en 4 dosis IV | 0,5 a 1g cada 6 horas IV | Máximo 4g diarios | |
| Meropenem | 120mg/kg/día en 3 dosis IV | 2g cad 8 horas IV | Máximo 6g diarios | |
| Piperacilina-Tazobactam | 350 a 450mg/kg/día en 4 dosis IV | 4.5g cada 6 horas IV | Máximo 16g diarios | |
| Ticarcilina-Clavulanato | 400mg/kg/día en 4 a 6 dosis IV | 3g cada 4 a 6 horas IV | Máximo 18g diarios | |
| Cefepime | 150mg/kg/día en 3 dosis IV | 2g cada 8 horas IV | Máximo 6g diarios | |
| Más uno de los siguientes | ||||
| Amikacina | 30 a 35mg/kg/día en 1 dosis IV | 30 a 35mg/kg/día en 1 dosis IV | Se ajusta de acuerdo a niveles plasmáticos y función renal | |
| Tobramicina | 10mg/kg/día en 1 dosis | 10 mg/kg/día en 1 dosis IV | Se ajusta de acuerdo a niveles plasmáticos y función renal | |
| Ciprofloxacino | 40mg/kg/día en 2 dosis oral 30mg/kg/día en 3 dosis IV | 750mg cada 12 horas oral 400mg cada 8 horas IV | Máximo 1.5g oral, 1.2g IV | |
| Colistin (colistimetato sódico) | 2.5 a 5mg/kg/día en 3 dosis IV | 2.5 a 5mg/kg/día en 3 dosis IV | Máximo 300mg diarios | |
| Haemophilus influenzae | Amoxicilina/ ácido clavulánico | 100mg/kg/día en 3 dosis oral | 1g cada 8 horas oral | |
| Cefotaxima | 200mg/kg/día en 4 dosis IV | 2g cada 8 horas IV | Máximo 8g diarios | |
3. Terapia de la infección crónica porPseudomonas aeruginosa
Una vez establecida la infección crónica, que es inerradicable, como vimos más arriba, el objetivo de la terapia ya no es obtener cultivos negativos, sino disminuir al máximo el número de colonias, para mantener la función pulmonar estable y evitar las exacerbaciones. La experiencia danesa en los 80's usando terapia endovenosa por 14 días con un betalactámico y un aminoglicósido, hospitalizando al paciente de manera electiva cada 3 meses, mostró un muy buen resultado mejorando ostensiblemente la sobrevida y la calidad de vida de los enfermos.
Actualmente este esquema ha sido reemplazado y superado con los antibióticos inhalados disponibles, que permiten tratamientos prolongados, en altas dosis, con mínima toxicidad y con excelente calidad de vida, al realizarse ambulatoriamente y sin alterar la actividad escolar o laboral habitual del paciente.
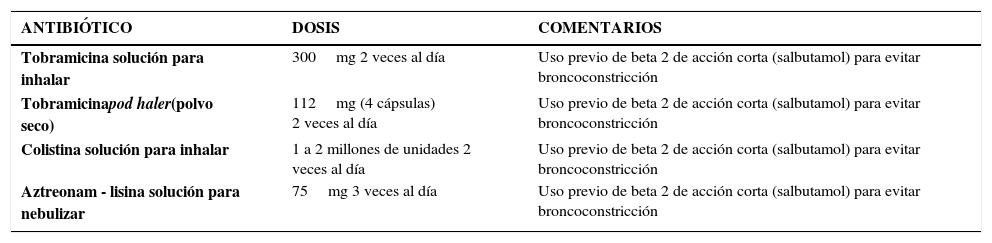
Inicialmente usamos los esquemas derivados de la evidencia médica, en que los antibióticos inhalados los indicábamos mes por medio (28 días con, 28 días sin medicamento), a permanencia. Actualmente nos hemos convencido de lo inadecuado que es dejar de administrar el antibiótico durante el mes de descanso, y nuestro esquema es usarlos permanentemente, sin pausas, idealmente alternando 2 o incluso 3 antibióticos inhalados 30,31. Los antibióticos inhalados y sus dosis aparecen en la Tabla 3.
ANTIBIÓTICOS PARA USO INHALATORIO EN FQ
| ANTIBIÓTICO | DOSIS | COMENTARIOS |
|---|---|---|
| Tobramicina solución para inhalar | 300mg 2 veces al día | Uso previo de beta 2 de acción corta (salbutamol) para evitar broncoconstricción |
| Tobramicinapod haler(polvo seco) | 112mg (4 cápsulas) 2 veces al día | Uso previo de beta 2 de acción corta (salbutamol) para evitar broncoconstricción |
| Colistina solución para inhalar | 1 a 2 millones de unidades 2 veces al día | Uso previo de beta 2 de acción corta (salbutamol) para evitar broncoconstricción |
| Aztreonam - lisina solución para nebulizar | 75mg 3 veces al día | Uso previo de beta 2 de acción corta (salbutamol) para evitar broncoconstricción |
4. Macrólidos, su aporte a la terapia
Derivado del efecto beneficioso de Azitromicina en la Panbronquiolitis obliterante, patología que se da preferentemente en Japón y que causa bronquiectasias e infección crónica por Pseudomonas aeruginosa, los estudios en FQ también han demostrado beneficio: en pacientes con infección crónica por Pseudomonas aeruginosa, mejora la función pulmonar, disminuye la frecuencia de las exacerbaciones y la necesidad de antibióticos 32. Incluso, en pacientes sin infección por P. aeruginosa, si bien no se observa cambio en la función pulmonar, disminuye significativamente la frecuencia de las exacerbaciones. Este efecto claramente no es antibiótico, dado que los macrólidos son inefectivos para P. aeruginosa, sino antiinflamatorio, probablemente bloqueando el “quorum sensing” y la producción del biofilm de alginato.
Inicialmente indicado en FQ con infección crónica por P. aeruginosa, actualmente muchos la indican en no infectados, pero con síntomas respiratorios persistentes o con disminución de la función pulmonar. Las dosis habituales son: 500mg día por medio en mayores de 40kg, 250mg día por medio en menores de 40kg y 5mg/kg día por medio en los más pequeños.
IV. MANTENER UN ESTADO NUTRICIONAL ÓPTIMO
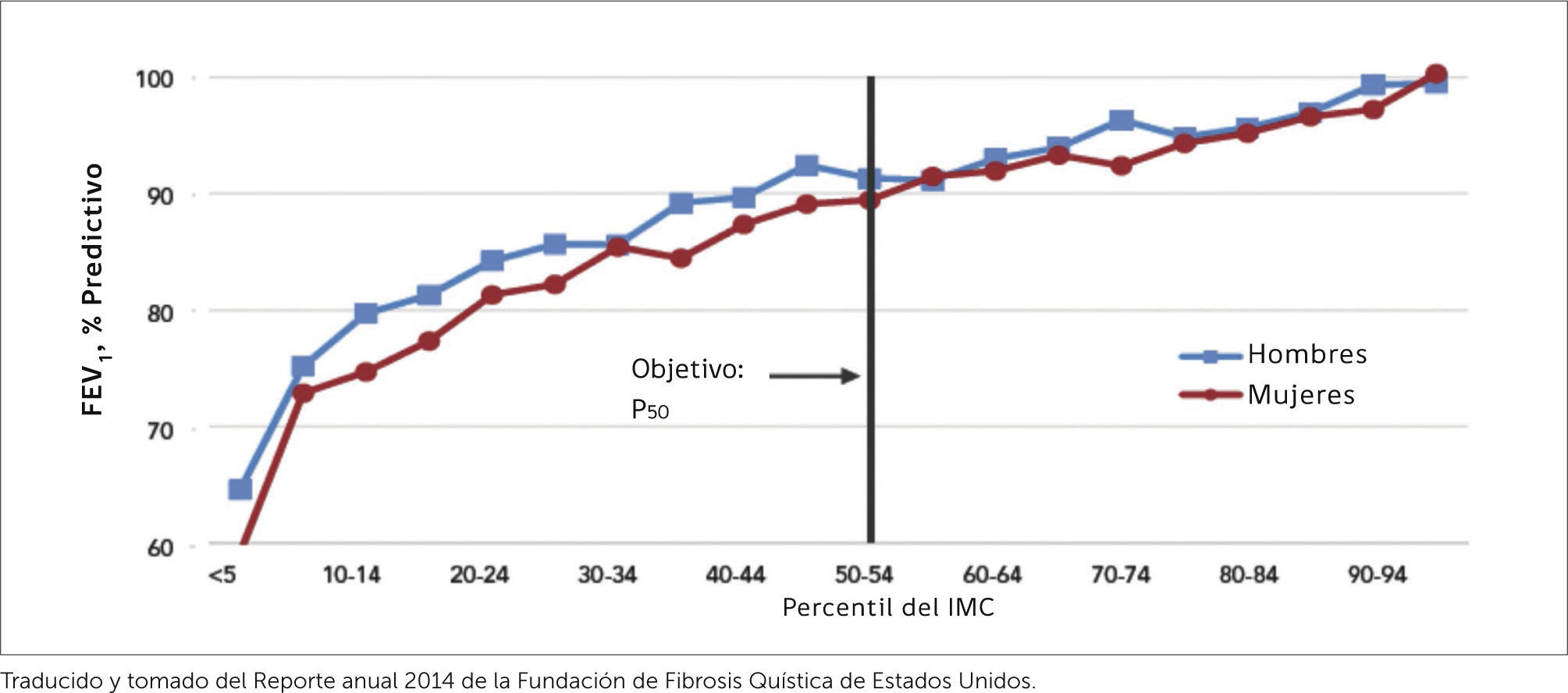
El estado nutricional es otro factor clave en el pronóstico de la FQ. Hay una correlación estrecha entre el índice de masa corporal (IMC) y la función pulmonar (VEF1), que es bidireccional, como se aprecia en la Figura 8, tomada de los datos del registro anual de la Fundación para la FQ de los Estados Unidos.

El objetivo del manejo nutricional es mantener un IMC sobre el percentil 50. Sobre el percentil 85 se considera sobrepeso. Entre el percentil 10 y el 50, se considera “riesgo nutricional” y bajo el percentil 10 se requiere de rehabilitación nutricional. En el menor de 2 años se utilizan los mismos criterios, usando como parámetro la relación peso/talla 28,29. En el adulto, el objetivo es mantener IMC sobre 22 en mujeres y sobre 23 en varones.
Para cumplir con el objetivo nutricional, se requiere:
- a)
Evaluación del IMC o de la relación peso/talla en todos los controles.
- b)
Terapia de reemplazo enzimático en la Insuficiencia Pancreática: el 80 a 85% de los casos de FQ se acompañan de falla pancreática, la que debe ser detectada desde el diagnóstico, y es la causa de la esteatorrea con malabsorción. Para el diagnóstico específico usamos actualmente los valores de la Elasta-1-fecal, que tiene la ventaja de requerir sólo una muestra de deposición y que no se altera por el uso de enzimas pancreáticas. Valores por debajo de 200 mcg/g de deposición sellan el diagnóstico de insuficiancia pancreática que debe ser tratada con enzimas de reemplazo. Se utilizan distintas formas comerciales, como Panzitrat o Creón, que viene en cápsulas con microesferas en su interior. Los niños mayores ingieren las cápsulas completas antes de cada alimentación, a los lactantes se les administran las microesferas.
Las dosis iniciales recomendadas se calculan en base al aporte de lipasa, y son: en menores de 4 años, 1000 unidades/kg/alimentación, en mayores de 4 años 500 unidades/kg/alimentación. Otro esquema es: 2000 unidades por 120ml de fórmula o de leche materna. Las dosis se regulan de acuerdo a la ganancia de peso y al aspecto de las deposiciones. No se debe superar el límite de 2500unidades/kg/alimentación o 10000 unidades/kg/día, para evitar el riesgo de colonopatía fibrosante 28,29.
- c)
Terapia de reemplazo de vitaminas liposolubles, A,D,E,K
Se utilizan preparados en gotas y comprimidos: para los menores de 1 año, 1ml /día, entre 1 y 3 años 2ml /día, entre 4 y 10 años, 1 comprimido/día y sobre los 11 años, 2 comprimidos/día.
- d)
Régimen hipercalórico, hiperproteico
Los pacientes de FQ requieren de las calorías suficientes para cumplir el objetivo del IMC o peso/talla sobre el percentil 50. En general, requieren el 150% de las calorías habituales para la edad.
- e)
Mantención de la salud ósea
A pesar de la administración de Vitamina D, la osteopenia con fracturas patológicas o deformidad de huesos aparecen con el tiempo. Por ello es importante monitorizar el nivel plasmático de Vitamina D anualmente, junto con los niveles de Calcio, Fósforo y Hormona paratiroidea, y evaluar la densitometría ósea a partir de los 8 años. Esta última se repite cada 2 años si es normal (Z score > -1)
- f)
Diabetes Mellitus relacionada a FQ (DMRFQ)
Alrededor del 15% de los adolescentes con FQ y el 50% de los adultos mayores de 30 años presentan DMRFQ, que es causa de caída del estado nutricional y de la función pulmonar. Por lo tanto, debe evaluarse su presencia con el Test de tolerancia a la glucosa anualmente, desde los 10 años de edad 31.
- g)
Enfermedad hepática de la FQ.
Entre el 20 al 40% de los pacientes de FQ desarrollan algún grado de disfunción hepática, de los cuales sólo una minoría llega a falla grave. Es importante evaluar anualmente las pruebas hepáticas y utilizar terapia de reemplazo de sales biliares cuando sea necesario 28–31.
V. MANEJO EN CENTRO ESPECIALIZADO MULTISISTÉMICO
Uno de los principales factores que explican la notable mejoría de la sobrevida media de los pacientes de FQ, es la atención en un Centro de FQ, donde el paciente es visto por un equipo multidisciplinario, especializado en la enfermedad, y donde encuentra solución a toda su problemática, de manera oportuna, fluída, integral, sistemática y bien registrada.
La formación de centros especializados permite además a los especialistas adquirir la experiencia, tan necesaria en el manejo de esta enfermedad, al concentrar un número adecuado de casos y complicaciones derivadas de ellos.
Los requisitos para formar un centro de FQ están estandarizados, e incluyen:
- 1.
Un equipo de especialistas en FQ: Neumólogo pediatra, gastroenterólogo pediátrico, nutriólogo, kinesiólogo, genetista, endocrinólogo pediátrico, neumólogo de adultos, asistente social. El Director del Centro es habitualmente el neumólogo y el sub director el gastroenterólogo, o viceversa.
- 2.
Un equipo de especialistas de apoyo: Otorrino, psiquiatra, psicólogo, cirujanos digestivo y torácico, infectólogo.
- 3.
Una enfermera coordinadora: Que hace el nexo de los especialistas con los pacientes, lleva el registro de los datos y facilita la atención integrada.
- 4.
Infraestructura y laboratorios: Test del sudor, Departamento de Imágenes, Laboratorio de Microbiología, Laboratorio de Función Pulmonar, Fibrobroncoscopía, Salas de Hospitalización y Unidad de Cuidados Intensivos.
- 5.
Un mínimo de 50 pacientes en control.
- 6.
Atención en Consulta ambulatoria integrada: Coordinada (con todos los especialistas juntos) y registrada estrictamente en una base de datos 31,33.
Es un hecho que el manejo de la FQ en centros especializados y multidisciplinarios donde el paciente recibe la atención integrada, basada en la evidencia, con protocolos estandarizados, además de la experiencia, calidez y oportunidad, ha cambiado la sobrevida, pero fundamentalmente la calidad de vida en nuestros pacientes.
El autor declara no tener conflictos de interés, en relación a este artículo.