



La fibrosis quística es una enfermedad multisistémica autosómica recesiva que ha presentado un ascenso de prevalencia en población hispana en las últimas décadas. Hace un poco más de 30 años se descubrió el gen regulador de la conductancia transmembrana de la fibrosis quística (CFTR). Hasta hoy, se han identificado 2106 mutaciones en el gen CFTR, 382 reconocidas como causantes de fibrosis quística. Según la consecuencia en la función de CFTR las mutaciones han sido clasificadas en 6 clases. Estos avances tecnológicos han permitido profundizar en el conocimiento de la estructura y función del CFTR, sus variantes y respuestas a terapias, lo que ha facultado que la medicina de precisión sea implementada en fibrosis quística. Con esto, se han desarrollado nuevos fármacos conocidos como moduladores. Dentro de ellos los más relevantes actualmente, al tener aplicación clínica, son los potenciadores y correctores. El uso conjunto en terapia dual y triple permite abarcar un mayor número de pacientes y muestra resultados superiores al mejorar la función pulmonar, disminuir exacerbaciones respiratorias y mejorar la calidad de vida de los pacientes. La fibrosis quística se ha convertido en un ejemplo de medicina de precisión, pero aún existen desafíos en cuanto a la accesibilidad a las terapias y ofrecer tratamiento a aquellos pacientes con mutaciones no incluidas en los fármacos disponibles o con respuesta inadecuada.
Cystic fibrosis is a hereditary multisystemic disease, that has presented a rise in prevalence in the Hispanic population in recent decades. A little more than 30 years ago, the cystic fibrosis transmembrane conductance regulator (CFTR) gene was discovered. To date, 2.106 mutations have been identified in the CFTR gene, 382 recognized as causing cystic fibrosis. According to the consequence in the CFTR function, the mutations have been classified into 6 classes. Those technological advances have made it possible to deepen our understanding of the structure and function of CFTR, its variants and responses to therapies, and in this way, they have allowed precision medicine to be implemented in cystic fibrosis. Therefore, new drugs, known as modulators, have been developed; enhancers and correctors being the most relevant among them due to their current clinical application. Their joint use in dual and triple therapy allows greater patient coverage and shows superior results by improving lung function, reducing respiratory exacerbations and improving quality of life. Cystic fibrosis has become an example of precision medicine, but there are still challenges in terms of accessibility to therapies and offering treatment to those patients with mutations not included in the available drugs or with an inadequate response.
La fibrosis quística es una enfermedad multisistémica autosómica recesiva. Ha sido descrita como la enfermedad hereditaria letal más frecuente en población blanca1, pero gracias al manejo terapéutico integral en centros especializados se ha convertido en una enfermedad crónica con una sobrevida media actual sobre los40 años en países desarrollados2. Su prevalencia es variable, se estima en 1 entre 3.500 a 4.000 recién nacidos en descendientes europeos2; en nuestro país3 se reporta una prevalencia de 1 entre 8.000 a 10.000 recién nacidos vivos. Si bien existen diferencias étnicas con una mayor proporción de población blanca afectada; en los últimos años se ha observado un ascenso de su prevalencia en población hispana4, con una mayor mortalidad comparada a no-hispanos en Estados Unidos5.
Las primeras descripciones de la enfermedad se remontan al siglo XVII, pero no fue hasta 1938 cuando fue reconocida como una entidad clínica distinta por Dorothy H. Andersen6. En 1989 se descubrió el gen regulador de la conductancia transmembrana de la fibrosis quística (CFTR, por sus siglas en inglés)7, ubicado en el brazo largo del cromosoma 7, el cual codifica para un canal aniónico activado por fosforilación y dependiente de AMPc, y que transporta cloro y bicarbonato a través de la membrana plasmática apical de células epiteliales. Modula además, la actividad de otros canales iónicos, como el canal de sodio epitelial (ENaC, por sus siglas en inglés)8. Al ser una enfermedad monogénica se han podido realizar múltiples estudios que relacionan genotipo y fenotipo profundizando el conocimiento y comprensión sobre la patología. Así mismo, se ha facilitado plantear tratamientos personalizados, ajustados al fenotipo del paciente. Por todos estos antecedentes, la fibrosis quística es considerada un ejemplo dentro de la medicina de precisión9.
2GenéticaLa fibrosis quística es una enfermedad monogénica, con una expresión clínica altamente compleja y variable. Al día de hoy se han identificado 2.106 variantes en el gen CFTR10, 466 de estas variantes han sido inscritas en el registro de traducción clínica y funcional de CFTR (CFTR2)11, entre ellas 382 variantes son causantes de fibrosis quística (es decir, patogénicas), 49 traducen manifestaciones clínicas variables, 11 presentan significado incierto (VUS, por sus siglas en inglés) y 24 de ellas no se asocian a fibrosis quística.
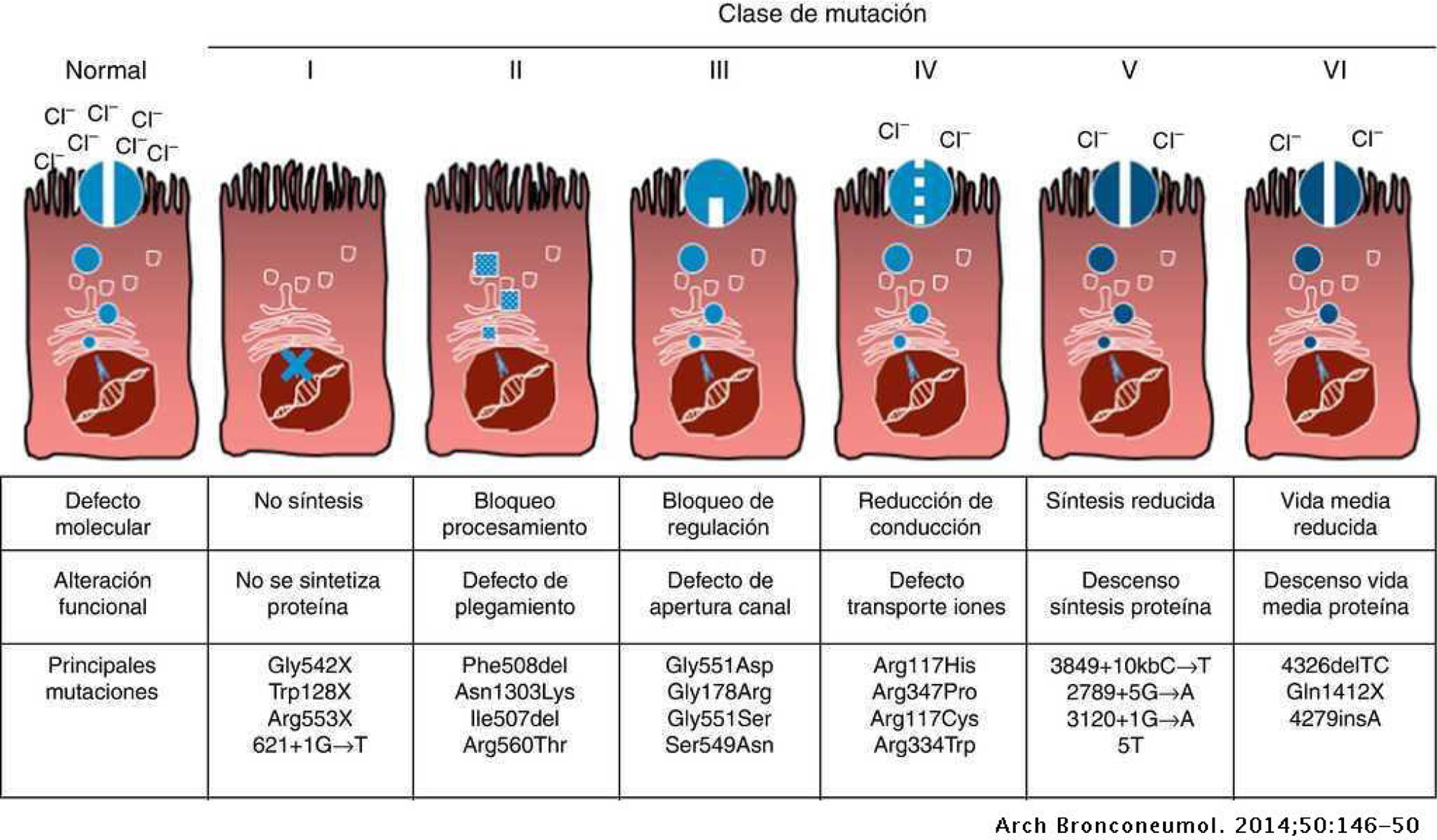
Según la consecuencia en la mutación del gen en la síntesis, estructura o función de la proteína CFTR, las mutaciones han sido clasificadas en grupos, siendo más frecuente encontrar la siguiente clasificación que las divide en 6 clases (Fig. 1)12,13:
.")
Tipos de mutaciones en la fibrosis quística. Reproducido de Quintana-Gallego et al. (Ref.13).
Clase I: Defecto de síntesis. Mutaciones que resultan en la pérdida total o parcial de la producción de una proteína funcional. Pueden deberse a la introducción de un codón de término prematuro, por cambios puntuales que provocan un desplazamiento del marco de lectura, como son las inserciones o deleciones de pequeñas regiones o exones, así como por la deleción parcial o completa del gen CFTR.
Clase II: Defecto de tráfico y procesamiento de la proteína debidos al mal plegamiento de ella. Estas mutaciones causan que la proteína sea retenida en el retículo endoplásmico y posteriormente degradada. La variante patogénica F508del (p.Phe508del), la más frecuente a nivel global, corresponde a esta clase.
Clase III: Defecto de regulación del canal CFTR. Es causada por ciertas mutaciones sin sentido (missense) en que la proteína se produce y se inserta en la membrana, pero es resistente a la activación y permanece cerrada.
Clase IV: Disminución de conductancia del canal. Es causada por ciertas mutaciones sin sentido que disminuyen la conductancia del canal con la consecuente alteración en el transporte de iones a través de este.
Clase V: Disminución de síntesis proteica. La reducción en el número total de proteínas CFTR puede deberse a alteraciones en el empalme de mRNA o cambios puntuales en la región promotora del gen.
Clase VI: Disminución de estabilidad proteica. Ciertas mutaciones reducen la estabilidad de CFTR en la membrana plasmática al aumentar su endocitosis o al reducir su retorno a la superficie celular.
Algunos autores incluyen dentro de la clasificación una séptima clase denominada “mutaciones irrecuperables”, la cual no es susceptible de manejo farmacológico y se debe a grandes deleciones del gen14,15.
Es importante mencionar que la asignación de cada mutación en esta clasificación no es del todo categórica, ya que una misma mutación puede tener características de más de una clase16.
Las mutaciones de clase IV, V y VI son conocidas como “mutaciones con función residual”, aquellos pacientes con al menos una mutación de este tipo presentan un debut más tardío de la enfermedad, con una menor severidad. Al contrario de las clases I, II y III, que corresponden a “mutaciones con mínima función” las cuales presentan escasa o ninguna función del CFTR y por lo tanto una enfermedad más severa8,14.
Gracias a los avances tecnológicos, que han permitido generar modelos computacionales y estudios en modelos celulares ex vivo, se ha podido profundizar en el conocimiento de la estructura y función del CFTR, sus variantes y respuestas a terapias. Así, la medicina de precisión ha sido implementada en fibrosis quística9.
3Medicina de precisiónEn medicina de precisión, la información molecular maximiza la exactitud con la que los pacientes son clasificados y tratados; entendiendo al individuo como una respuesta a la interacción entre el ambiente, estilo de vida, adherencia al tratamiento y factores genéticos. Esto ha permitido la creación de nuevos modelos terapéuticos al ajustar fármacos convencionales a un tipo y dosis individualizada al paciente, y el desarrollo de nuevos fármacos conocidos como moduladores8,9.
4ModuladoresLos moduladores tienen la habilidad de mejorar o incluso restaurar la expresión, función y estabilidad de un CFTR defectuoso8. Se han clasificado en 5 grupos según su efecto sobre las mutaciones de CFTR: potenciadores, correctores, estabilizadores, agentes de lectura y amplificadores.
4.1PotenciadoresCorresponden a moléculas que restauran o aumentan la probabilidad de apertura del canal, permitiendo la conductancia (mutaciones tipo III y IV)8.
Ivacaftor (VX-770; Farmacéutica Vertex) es un potenciador que restaura parcialmente la actividad de CFTR al unirse a la interfaz entre dominios de transmembrana de CFTR17 y promueve el desacople entre el ciclo de activación del canal y la hidrolisis de ATP, manteniendo el canal abierto por más tiempo18; pero su mecanismo de acción no está del todo dilucidado.
Fue el primer modulador aprobado para el uso en pacientes con fibrosis quística en el año 2012 tanto por la Administración de Medicamentos y Alimentos de Estados Unidos (FDA) y la Agencia Europea de Medicamentos (EMA), en pacientes sobre 6 años con al menos una mutación G551D (p.Gly551Asp) (mutación clase III para el cual fue diseñado inicialmente, y el cual se ha encontrado con una frecuencia alélica de 2,1% en las bases de datos de CFTR2)11,19. Desde entonces son múltiples los beneficios clínicos a corto y largo plazo que se han reportado con este potenciador. Ivacaftor reduce los valores de cloro en el sudor en 48,1mmol/L alcanzado niveles bajo el corte utilizado para el diagnóstico de 60mmol/L 20, también se observó un aumento de 10,8 puntos porcentuales en el porcentaje del predicho de VEF1 a las 24 semanas de seguimiento20 asociado a enlentecimiento en el deterioro de la función pulmonar20,21. Además, se ha descrito que ivacaftor mejora el clearance mucociliar22, reduce el número de exacerbaciones respiratorias hasta en un 55%20,21, disminuye el número de días de uso de antibiótico23, el número de hospitalizaciones21,24; y disminuye la detección de Pseudomona aeruginosa22. Desde el punto de vista nutricional, se ha descrito un aumento promedio de 2,7 Kg. en pacientes adultos20, mejora el índice de masa corporal tanto en niños como adultos25, y mejora el tiempo de ejercicio26 y la calidad de vida27.
En años recientes la aplicación clínica de ivacaftor se ha ampliado, esto apoyado por evidencia que reporta beneficios de su uso en otras mutaciones con función residual23,28–30, y la reciente publicación de estudios sobre la seguridad y eficacia de ivacaftor en lactantes desde los 4 meses de vida al mejorar niveles de cloro en sudor, y marcadores de función pancreática31,32. Actualmente se encuentra aprobado para su uso en pacientes desde los 4 meses de edad y con al menos una copia de alguna de las 97 mutaciones especificas33, estas mutaciones corresponden a clase III y IV. Nuevos potenciadores están siendo estudiados y se encuentran en fase experimental y estudios clínicos8.
4.2CorrectoresComo se mencionó previamente, la mutación más frecuente es la F508del, mutación clase II. Los correctores son compuestos que corrigen el tráfico de la proteína que presenta defectos en el plegamiento, usualmente mejoran la estabilidad conformacional de la proteína durante el proceso de plegado en el retículo endoplasmático. Los correctores permiten el paso de la proteína a la membrana celular al unirse a la proteína mutada directamente (chaperones), o de manera indirecta al modular su interacción con la homeostasis proteica (reguladores de la proteostasis)8.
Lumacaftor (VX-809; Farmacéutica Vertex) es un corrector de primera generación; su uso conjunto con ivacaftor mostró un aumento de 3,6 puntos porcentuales en el porcentaje del predicho de VEF1 en pacientes mayores de 18 años con mutación F508del homocigota34. Posterior a esto, una revisión Cochrane35 reportó un aumento de 2,8 a 3,3 puntos porcentuales en el porcentaje del predicho de VEF1 con terapia dual y dosis de 400 y 600mg de lumacaftor respectivamente a los 6 meses de seguimiento.
La asociación lumacaftor/ivacaftor disminuye además, el número de exacerbaciones en un 30-39%, logra una mejoría en IMC, enlentecimiento del deterioro de la función pulmonar y mejoría en función pulmonar evaluada por LCI2,5, disminución en niveles de cloro en sudor y mejoría en marcadores de función pancreática25,36–39. Cabe destacar que la respuesta en otras mutaciones de clase II y en pacientes con mutación F508del heterocigota ha sido variable y no ha demostrado los mismos beneficios8.
En 2015, la FDA y EMA autorizaron el uso conjunto de lumacaftor/ivacaftor (Orkambi®; Farmacéutica Vertex) en pacientes homocigotos de F508del y mayores de 12 años y desde el 2018 está autorizado su uso en mayores de 2 años homocigotos de F508del en base a estudios que mostraron seguridad en este grupo de edad37,39.
Lumacaftor gatilla la activación del citocromo P450 lo que resulta en una concentración plasmática reducida de ivacaftor8. Como efectos adversos se han reportado tos, aumento de secreciones, hemoptisis y disnea inicial36,38.
Tezacaftor (VX-661; Farmacéutica Vertex) es un corrector de segunda generación con mejores propiedades farmacocinéticas y menos efectos adversos8. Aprobado en 2018 por la FDA y EMA en combinación tezacaftor/ivacaftor (Symdeko® o Symkevi® Farmacéutica Vertex) en pacientes mayores de 12 años con mutación homocigota de F508del y heterocigota con una mutación F508del más una mutación con función residual. Al año siguiente se autorizó su uso en pacientes mayores de 6 años19. Los estudios que fundamentan su uso han demostrado en pacientes homocigotos F508del aumento de 3,4 puntos porcentuales en el porcentaje del predicho de VEF1, disminución de exacerbaciones respiratorias en un 35% y un descenso en nivel de cloro en sudor comparado con placebo de 8mml/L40-42. En pacientes heterocigotos con una copia de F508del y de G551D se observó un aumento de 4,6 puntos porcentuales en el porcentaje del predicho de VEF1 y un descenso en el nivel de cloro en sudor comparado con placebo41. En pacientes heterocigotos con una copia F508del más una mutación con función residual tezacaftor/ivacaftor aumentó 6,8 puntos porcentuales en el porcentaje del predicho de VEF1 (ivacaftor monoterapia aumentó 4,7 puntos porcentuales) comparado con placebo, también se observó mejora en cuestionarios de calidad de vida42. Un estudio en 70 niños entre 6 a 11 años con mutación homocigota y heterocigota de F508del más una mutación con función residual demostró la seguridad del fármaco y una disminución en niveles de cloro en sudor y mejora en calidad de vida; función pulmonar y crecimiento se mantuvieron estables en rangos normales43.
La combinación tezacaftor/ivacaftor actualmente se encuentra aprobado para su uso en pacientes desde los 6 años de edad homocigotos de F508del o con al menos una copia de alguna de 154 mutaciones especificas44.
Considerando los resultados discretos de las combinaciones de ivacaftor con lumacaftor y luego tezacaftor, se desarrollaron nuevos correctores; bamocaftor (VX-659) y elexacaftor (VX-445) han mostrado un buen perfil de seguridad para su uso a largo plazo. Se planteó el uso sinérgico de tezacaftor/ivacaftor más un segundo corrector con distinto mecanismo de acción para potenciar la respuesta clínica8. Es así como en 2019 se autoriza el uso de Trikafta™ de farmacéutica Vertex que combina tezacaftor/ivacaftor/elexacaftor para mayores de 12 años con al menos una mutación F508del, lo que brinda una nueva opción terapéutica a cerca del 90% de los pacientes con fibrosis quística19. En el caso específico de Chile, de acuerdo a nuestro tipo de mutaciones, este medicamento beneficiaría entre 75% a 80% de los pacientes con fibrosis quística3. Trikafta™ logró un aumento de 11,8 y 13,8 puntos porcentuales en el porcentaje del predicho de VEF1 en pacientes mayores de 18 años homocigotos para F508del y heterocigotos F508del más una mutación con función residual respectivamente, además, ambos grupos presentaron descenso en los niveles de cloro en sudor y mejor puntaje en cuestionarios de calidad de vida45. En un estudio de 403 pacientes mayores de 12 años heterocigotos F508del más una mutación con función residual por 24 semanas se observó un aumento de 14,3 puntos porcentuales en el porcentaje del predicho de VEF1, disminución en exacerbaciones respiratorias en un 63%, disminución en niveles de cloro en sudor de 41,8mmol/l y mejor calidad de vida comparado con placebo46. Zemanick et al.47 evaluaron la seguridad y eficacia de Trikafta™ en 66 niños de 6 a 11 años con al menos una copia de F508del, los efectos adversos reportados fueron de leves a moderados, a las 24 semanas de seguimiento se reportó una mejor función pulmonar con un aumento de 10,2 puntos porcentuales en el porcentaje del predicho de VEF1 y un descenso de 1,71 puntos en LCI2,5 comparado con mediciones basales; además de mayor puntaje en encuestas de calidad de vida, mejoría del IMC y disminución en valores de cloro en sudor en 60,9mmol/L. Estos hallazgos permitieron la aprobación de Trikafta™ para pacientes desde los seis años en junio 2021 con al menos una copia de F508del o al menos una copia de 177 mutaciones específicas48.
4.3EstabilizadoresLos estabilizadores son agentes que anclan el CFTR a la membrana celular, previniendo su eliminación y degradación por lisosomas. Los pacientes con mutaciones tipo VI podrían beneficiarse con estas terapias. In vitro son varios los agentes que han mostrado modificar la inestabilidad proteica de CFTR. Cavosonstat (N91115; Nivalis) fue el primer estabilizador evaluado en estudios clínicos y si bien inicialmente no evidencio problemas de seguridad y una reducción de concentración de cloro en sudor en pacientes homocigotos F508del8, ensayos clínicos de fase II no demostraron beneficio adicional en función pulmonar ni en niveles de cloro en sudor, tanto en su uso como monoterapia como al combinar con lumacaftor/ivacaftor ni con ivacaftor35.
4.4Agentes de LecturaSon compuestos que inducen una sobre-lectura ribosómica de un codón de terminación prematura, lo que permite la incorporación de un aminoácido en ese lugar y, continuar la traducción al final normal de la transcripción; por lo tanto, están dirigidos a mutaciones clase I. Las primeras propiedades de agente de lectura fueron encontradas en aminoglucósidos (gentamicina), pero fue descartado su uso clínico debido al riesgo de nefro y ototoxicidad8.
Ataluren (PTC124; Terapéutica PTC) demostró restaurar la expresión y función de CFTR en modelos animales, pero no exhibió beneficios en función pulmonar ni número de exacerbaciones en estudios fase 349,50.
ELX-02 (NB124; Farmacéutica Eloxx) ha demostrado in vitro e in vivo producir proteínas normales y funcionales en mutaciones sin sentido51. En estudios fase 1 se ha estudiado la farmacocinética y seguridad de ELX-02 en sujetos sanos, con eventos adversos leves a moderados reportados52,53; esto dio el paso para iniciar el estudio del fármaco en pacientes con mutaciones sin sentido, actualmente existen dos estudios fase 2 en pacientes con fibrosis quística en reclutamiento (NCT04126473, NCT04135495).
4.5AmplificadoresCompuestos que aumentan la expresión de mRNA de CFTR y por lo tanto la biosíntesis de la proteína CFTR. Está orientado al tratamiento de pacientes con mutaciones tipo V.
PTI-428 (Nesolicaftor; Terapéutica Proteostasis) aumenta de forma selectiva la expresión de proteína CFTR inmadura sin provocar alteración en la expresión de genes de respuesta al estrés celular. En un estudio clínico fase 1/2 (NCT03500263) la combinación triple de PTI 428, 801 y 808 mejoró 8% el porcentaje del predicho de VEF1 en pacientes adultos homocigotos F508del en seguimiento de 28 días. Existen otros estudios clínicos actualmente en desarrollo para evaluar su efectividad y seguridad8
5Terapia génicaLuego de la identificación del gen CFTR un número creciente de estrategias en terapia génica fueron probadas para intentar curar la fibrosis quística sin éxito. Los recientes avances en ingeniería genética y el desarrollo de herramientas biotecnológicas más eficaces han reavivado el interés por esta alternativa de manejo. Es así como en modelos animales y celulares se han reportado resultados favorables con técnicas de terapia génica. Mediante la técnica de reparación por extremos no homólogos, usando CRISPR-nucleasas, una familia de secuencias de DNA/RNA y una proteína que se dirige a zonas elegidas de DNA y lo edita, se logró una reparación y recuperación de la función de CFTR en modelos celulares con mutaciones específicas de corte y empalme (splicing)54. Esta nueva tecnología es prometedora ya que permite generar nuevas herramientas experimentales y terapéuticas para una amplia variedad de mutaciones de CFTR55.
6ConclusionesEn los últimos años importantes avances tecnológicos han permito profundizar el conocimiento genético de diversas enfermedades, y en el caso de la fibrosis quística, ofrecer terapias nuevas y más específicas que han significado grandes cambios en la vida de muchos pacientes, mejorando distintos aspectos que incluyen función pulmonar, estado nutricional, número de exacerbaciones y hospitalizaciones y mejoría en la calidad de vida.
Un punto de tope importante en las terapias que actualmente existen es su accesibilidad, ya sea por sus altos costos o que no se encuentren disponibles, como es el caso en nuestro país.
Aún queda mucho camino por recorrer en las posibilidades terapéuticas individualizadas para pacientes con fibrosis quísticas. Las terapias existentes si bien agrupan a un número importante de pacientes aún dejan fuera aquellos que son portadores de mutaciones tipo I y tipo VII, o quienes que por eventos adversos no pueden utilizar estos tratamientos de por vida. Estudios in vitro con líneas celulares derivadas de pacientes con mutaciones raras y poco frecuentes de CFTR dan una alternativa para evaluar y predecir una respuesta terapéutica a nivel individual.
Declaración de conflicto de interésAmbas autoras declaran no presentar conflicto de interés.