



La piel posee un doble origen embriológico ectomesodérmico, por lo que se relaciona con todos los órganos y sistemas del organismo. Es por esto que la piel puede reflejar alteraciones sistémicas de todo tipo, existiendo algunos marcadores dermatológicos bien reconocidos que pueden preceder, acompañar o seguir al diagnóstico de una enfermedad sistémica. La fácil accesibilidad de la piel para toma de muestra (cultivos, biopsias) facilita el estudio a cabalidad. El integrar los hallazgos clínicos con las lesiones de la piel, es una tarea difícil, que tanto dermatólogos como otros especialistas deben poner especial atención.
Al estudiar el organismo humano desde el punto de vista de los diferentes sistemas se puede observar cómo en cada caso existen lesiones cutáneas que están íntimamente ligadas a estas patologías. En algunos casos estas lesiones son parte de la enfermedad o en otros casos son producto de la enfermedad.
The skin has a double embryological origin therefore relates to all organs and body systems. For this reason, the skin may reflect systemic changes of all kinds, with some well-recognized dermatological markers that may precede, accompany or follow the diagnosis of systemic disease. The easy accessibility of skin samples (for cultures, biopsies) facilitates research. Integrating clinical findings and skin lesions is a difficult task, for both dermatologists and other specialists, close attention is necessary.
In studying the human organism from the viewpoint of the different systems you can see how in each case there are skin lesions that are closely related to these pathologies. In some cases these lesions are part of the disease or in other cases are caused by the disease.
Existen distintos trastornos del aparato digestivo que producen alteraciones cutáneas reconocibles como parte de su espectro clínico. Dentro de ellas:
a)Asociadas a hemorragias digestivas1. Pseudoxantoma elástico: es un trastorno hereditario muy poco frecuente caracterizado por alteración del tejido elástico por calcificación progresiva y elastorrexis (fragmentación) de las fibras elásticas de la dermis reticular, principalmente de la piel, retina y vasos sanguíneos. Las lesiones cutáneas aparecen entre la segunda y tercera década de la vida y se caracterizan por la presencia de placas amarillentas, de superficie “en empedrado o piel de gallina”, en cuello, pliegues axilar e inguinal y periumbilical. A nivel ocular se observan estrías angioides en la retina. La alteración de las fibras elásticas de los vasos de mediano calibre causa isquemia coronaria, accidentes vasculares cerebrales, hipertensión severa, además de hemorragias digestivas (1).
2. Telangiectasia hemorrágica hereditaria o Sindrome de Rendu-Osler Weber: es un trastorno autosómico dominante que se caracteriza por una displasia vascular multisistémica, produce capilares ectásicos mucocutáneos y viscerales. Suele manifestarse en la adolescencia con lesiones mucosas de la nariz y la boca, que causan epistaxis repetidas en un 90% de los pacientes. En la tercera década de la vida, las telangiectasias se extienden a cara, punta de los dedos y otras localizaciones. Las hemorragias gastrointestinales suelen comenzar entre la cuarta y sexta década de la vida. Pueden presentar fístulas arteriovenosas pulmonares. Los pacientes pueden sufrir anemia crónica e insuficiencia cardíaca (2).
b)Asociadas a poliposis1. Sindrome de Gardner: Autosómico dominante. Se caracteriza por la presencia de pólipos adenomatosos principalmente colorrectales que pueden malignizar. Además existen diversos tumores benignos cutáneos como quistes epidermoides, fibromas subcutáneos, lipomas además de osteomas (3, 4).
2. Sindrome de Peutz-Jeghers: Autosómico dominante. Los pólipos se localizan a nivel gastrointestinal, son hamartomatosos por malformación del tejido conjuntivo, poseen rara degeneración neoplásica, dando con más frecuencia problemas mecánicos (torsión, sangrado, oclusión). Las lesiones mucocutáneas son máculas pigmentadas lentiginosas, marrones o azuladas, en labios, mucosa oral y punta de dedos (5).
c)Asociadas a malabsorción1. acrodermatitis enteropática: enfermedad caracterizada por malabsorción de zinc. Se transmite de forma autosómica recesiva. Se han descrito formas adquiridas en pacientes que reciben nutrición parenteral total con deficiencia de zinc, alcoholicos, desnutrición crónica. Las manifestaciones cutáneas son polimorfas, observándose vesículo-ampollas, pústulas, eccemas acrales, periorales y perirrectales. En cara y pliegues flexores se observan placas eritematosas descamativas. Hay alopecia y onicodistrofia. El paciente presenta diarrea, irritabilidad y alteración de la curva de crecimiento. Los síntomas comienzan entre el primer mes y los 18 meses, tras el cambio de la leche materna a la leche de vaca, debido a la ausencia del factor transportador de zinc. La medición de la concentración plasmática de zinc permite hacer el diagnóstico (6, 7).
2. Dermatitis herpetiforme: es una manifestación cutánea de la enfermedad celíaca. Se producen pápulas y vesículas eritematosas agrupadas en las superficies extensoras de antebrazos, codos, rodillas, glúteos. O solo prurito... El estudio histológico demuestra una ampolla a nivel de la membrana basal, con neutrófilos intrapapilares y la inmunofluorescencia detecta el depósito de IgA granular en las prolongaciones papilares de la dermis. Más del 85% de los pacientes muestra inflamación intestinal en la biopsia yeyunal. Los signos y síntomas concomitantes de malabsorción son proporcionales a la gravedad de la enteropatía por gluten (8).
d)Asociadas a enfermedad inflamatoria intestinal (EII): Enfermedad de Crohn y colitis ulcerosa1. Manifestaciones específicas: Se denominan así porque se caracterizan histopatológicamente por la presencia de granulomas idénticos a los encontrados en el intestino. Son más frecuentes en la enfermedad de Crohn. Pueden encontrarse en áreas periorificiales (Crohn oral y perianal), alrededor de las fístulas o a distancia (Crohn “metastásico”) (9-11).
2. Manifestaciones inespecíficas o reactivas: Su curso suele coincidir con una exacerbación de la enfermedad digestiva.
2.1. Aftas orales: Úlceras superficiales de la mucosa oral, recubiertas por una membrana de fibrina central, de color blanco-grisáceo, y rodeadas de un halo inflamatorio. Son más frecuentes en el curso de la colitis ulcerosa. Las lesiones son idénticas a las aftas orales idiopáticas y a las aftas observadas en la enfermedad de Behçet.
2.2. Eritema nodoso: Se caracteriza por nódulos subcutáneos eritematosos, pretibiales, dolorosos a la palpación, que histopatológicamente corresponden a una paniculitis septal. Esta dermatosis puede verse también asociada a infecciones, a sarcoidosis, a fármacos, al embarazo, a neoplasias o de forma idiopática.
2.3. Dermatosis neutrofílicas: Enfermedades que aparecen como reacción a diferentes estímulos (Enfermedad intestinal inflamatoria crónica, enfermedad de Behçet, síndromes mieloproliferativos) y que se caracterizan por la presencia de leucocitos polimorfonucleares en la piel (12).
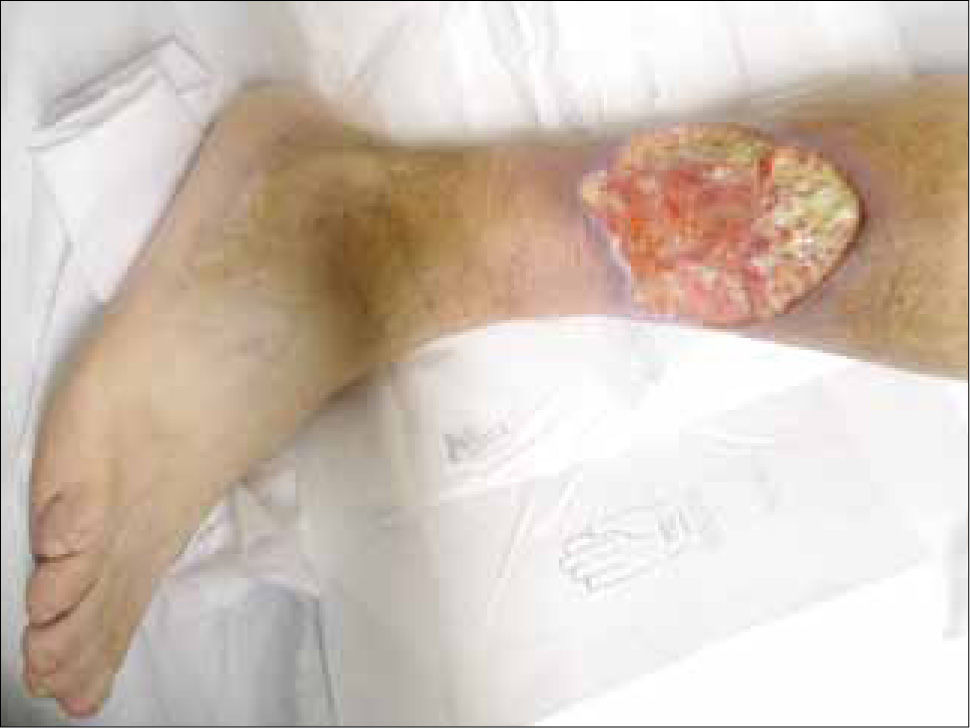
2.3.1. Pioderma gangrenoso: El 1-5% de los pacientes con EII desarrollan pioderma gangrenoso y a su vez, la EII es la causa más frecuente de pioderma gangrenoso, siendo la responsable del 15-25% de los casos. Clínicamente hay ulceraciones intensamente dolorosas, de crecimiento rápido, localizadas habitualmente en miembros inferiores, de bordes socavados de color violáceo, que a la presión dejan salir un contenido purulento. Suele asociarse a colitis ulcerosa y muy rara vez a enfermedad de Crohn. Como otras dermatosis neutrofílicas presenta “fenómeno de patergia” (tendencia a desarrollar un cuadro de morfología similar al puncionar la piel). En el caso del pioderma gangrenoso, puede verse alrededor de los orificios de colostomía, empleada como tratamiento de estos enfemos (Figura 1).

2.3.2. Síndrome de Sweet: También denominado “dermatosis aguda neutrofílica y febril”, de inicio agudo, con fiebre, compromiso del estado general y leucocitosis con neutrofilia en sangre periférica. Las lesiones cutáneas son pápulas y placas eritematosas, edematosas, de aspecto vesiculoso en superficie, dolorosas, que predominan en la mitad superior del cuerpo. Puede verse también asociado a infecciones respiratorias, fármacos, neoplasias (especialmente leucemias mieloides aguda y crónica).
2Manifestaciones cutáneas asociadas a hepatopatíasLas hepatopatías se asocian a diversos signos cutáneos, aunque ninguno de ellos es específico. Los cambios cutáneos observados en las hepatopatías pueden deberse a alteraciones primarias del hígado; a enfermedades cutáneas con anomalías hepáticas asociadas; y a una gran variedad de trastornos que cursan con cambios en muchos órganos, incluidos el hígado y la piel (13).
1. Cirrosis hepática: Alteración irreversible de su arquitectura, consistente en fibrosis hepática. Su principal causa es el alcohol, puede deberse también a fármacos, infecciones virales (hepatitis C), obstrucción biliar. Hay ictericia, prurito, arañas vasculares, eritema palmar, dilataciones venosas abdominales por hipertensión portal, uñas de Terry (blancas y opacas con extremo distal rosado) y pérdida del vello pectoral, axilar y púbico. Si se trata de cirrosis biliar primaria hay además prurito incoercible, ictericia, hiperpigmentación postinflamatoria y múltiples xantomas, estos últimos como consecuencia de la hiperlipidemia asociada.
2. Hemocromatosis: Trastorno autosómico recesivo que se caracteriza por hiperpigmentación, diabetes mellitus y cirrosis hepática. Existe un defecto básico en el metabolismo del fierro que favorece su absorción intestinal y su depósito principalmente en piel (más llamativo en zonas fotoexpuestas, genitales, pliegues y areolas), además de otros órganos. Se conoce también como "Diabetes bronceada" (14).
3. Enfermedad de Wilson: Trastorno autosómico recesivo muy poco frecuente, asociado a cirrosis, aunque sus manifestaciones clínicas se deben a una acumulación excesiva de cobre en numerosos tejidos. Existe una triada característica que consiste en degeneración de ganglios basales, cirrosis hepática y pigmentación patognomónica de los bordes corneales (anillos de Kayser-Fleischer) (15).
3Manifestaciones cutáneas asociadas a pancreatopatíasLas manifestaciones cutáneas de las pancreatopatías pueden deberse a las funciones exocrinas/endocrinas del páncreas o a los carcinomas pancreáticos (16).
1.Pancreatitis: Las manifestaciones cutáneas son más frecuentes en las pancreatitis agudas y son fundamentalemente de 2 tipos: púrpura y paniculitis. Por extravasación subcutánea de líquido hemorrágico peritoneal, pueden observarse los signos de Turner y Cullen, los cuales corresponden a equimosis en el costado y en la región periumbilical respectivamente. La paniculitis pancreática puede aparecer tanto en la pancreatitis como en el carcinoma pancreático, y puede manifestarse en forma de necrosis adiposa subcutánea o nodular. No se conoce bien su etiología, la mayoría de las teorías se centran en la acción enzimática de lipasas que el páncreas dañado libera a la circulación, y que degradan el tejido subcutáneo. Clínicamente se aprecian nódulos inflamatorios en muslos, parte inferior piernas, también en brazos, glúteos o tronco.
2. Síndrome del glucagonoma: Corresponde a una pancreatopatía endocrina no diabética. La erupción cutánea característica es el eritema necrolítico migratorio, y en la mayoría de los casos representa un marcador cutáneo de un tumor de células α del páncreas. La erupción cutánea suele ser extendida y presenta una distribución típica, afectando principalmente las zonas perioral e intertriginosas, especialmente periné. La necrosis superficial de la epidermis produce una dermatosis erosiva y en ocasiones vesículoampollar, con formación de costras y desprendimiento final de la piel. La base de las lesiones suele ser muy eritematosa, y los bordes son anulares o serpiginosos (16).
4Manifestaciones cutáneas asociadas a nefropatíasLas manifestaciones cutáneas de las enfermedades renales son frecuentes, sobre todo en el contexto de una insuficiencia renal crónica (IRC).
a) Prurito: es la manifestación dematológica más frecuente de la IRC. La causa fundamental es la menor depuración de las moléculas que producen prurito, pero no se ha identificado la molécula culpable. Los pacientes con insuficiencia renal poseen una piel seca y deshidratada con palidez cutáneo-mucosa (por la anemia subyacente) e hiperpigmentación en zonas fotoexpuestas. Además, con frecuencia padecen de prurito muy intenso que no responde al tratamiento con antihistamínicos sistémicos ni corticoides tópicos. La fototerapia con radiación ultravioleta UVB es útil en estos pacientes (17).
b) Dermatosis perforantes adquiridas: Combinan características de distintos tipos de dermatosis perforantes (con eliminación transepidérmica o folicular de diversas sustancias) como la foliculitis perforante, la enfermedad de Kyrle y la colagenosis perforante reactiva. En general, las lesiones comienzan pocos meses después de iniciar la diálisis. Se asocian a prurito renal y a diabetes mellitus. Consisten en pápulas o nódulos que con frecuencia poseen un tapón queratósico central y pueden coalescer formando placas verrucosas. Se localizan en las zonas flexoras de las extremidades, pero también en tronco, cuello, cara y, más raramente, cuero cabelludo. Pueden tratarse con queratólíticos, UVB o individualmente con crioterapia. Pueden autoinvolucionar si se suspende la hemodiálisis, por ejemplo, después del trasplante renal (18).
c) Fibrosis sistémica nefrogénica (FSN): enfermedad que afecta piel y tejido subcutáneo y se asemeja a la esclerodermia y al escleredema. Se desconoce la etiología exacta, es un proceso que sólo afecta a pacientes con nefropatía, la mayoría en diálisis. Hace poco se ha descrito que la FSN aparece en los pacientes a los que se les ha realizado resonancia magnética con gadolinio como medio de contraste. El gadolinio libre disociado en los tejidos de pacientes con IRC podría ser un objetivo para los fibrocitos circulantes. La mayoría son pacientes de mediana edad, puede afectar niños y ancianos. Clínicamente se produce un engrosamiento simétrico de la piel de extremidades, y en ocasiones del tronco, aunque por lo general, respeta la cara. La zona más frecuentemente afectada es la región desde los tobillos a la mitad de los muslos, y al principio puede confundirse con una celulitis. Al inicio las lesiones son eritematosas y después se vuelven "leñosas" con una superficie en piel de naranja. El engrosamiento progresivo de la piel sobre las articulaciones limita la movilidad. No existe ninguna medida individual que mejore la evolución de la FSN. Estudios han demostrado eficacia de la plasmaféresis, Re PUVA (19).
d) Calcifilaxia: Es una enfermedad infrecuente y muy grave que se produce por calcificación de la capa media de las arteriolas y de las pequeñas arterias de la dermis e hipodermis, que ocurre generalmente en el contexto una IRC. Aunque su etiopatogenia no está completamente clara, en un 80% de los casos existe hiperparatiroidismo y/o un producto calcio-fósforo elevado. Clínicamente, se caracteriza por la aparición súbita de lesiones cutáneas en forma de máculas violáceas dolorosas que adoptan un patrón retiforme (tipo lívedo reticular), que tienden a necrosarse parcialmente, dejando úlceras recubiertas por escaras necróticas. Se localizan con mayor frecuencia en extremidades inferiores, y en regiones con gran cantidad de grasa subcutánea y por lo general, respeta la cara. Es una enfermedad muy resistente al tratamiento y con muy mal pronóstico (17).
5Enfermedades suprarrenales y pielDentro de las enfermedades suprarrenales encontramos al Sindrome de Cushing producido por el exceso crónico de glucocorticoides y la Enfermedad de Addison causada por insuficiencia crónica primaria de la glándula (20).
a) Sindrome de Cushing: El exceso de corticoides se ve causado por hipersecreción pituitaria de hormona adreno-corticotrópica (ACTH) la cual corresponde específicamente a la Enfermedad de Cushing, la secreción ectópica de ACTH por tumores no hipofisiarios, hipersecreción adrenal o administración exógena de corticoides. Los cambios dermatológicos traducen una alteración de la distribución subcutánea de la grasa, lo que clínicamente se manifiesta como "cara de luna", "cuello de búfalo", depósito graso en la cintura pelviana y reducción de la grasa en brazos y piernas. Se ve atrofia cutánea generalizada con disminución de los componentes epidérmicos y dérmicos, estrías, fragilidad cutánea, púrpura (por reducción del soporte conectivo) y enlentecimiento en la curación de heridas. Es posible apreciar también acné esteroidal e hirsutismo.
b) Enfermedad de Addison: La insuficiencia suprarrenal puede deberse a una adrenalitis autoinmune y menos frecuente a infección, hemorragia o neoplasia. Las manifestaciones cutáneas son principalmente hiperpigmentación (por efecto de la hormona melanocitoestimulante) en zonas fotoexpuestas, sitios de trauma, axilas, periné, areolas, pliegues palmares, membranas mucosas y uñas. Puede haber pérdida aumentada de sal en el sudor.
6Manifestaciones cutáneas de los trastornos endocrinosa) Manifestaciones cutáneas de la Diabetes: Además de una mayor tendencia a infecciones cutáneas y al desarrollo del denominado “pie diabético” (úlceras crónicas secundarias a la inmunode-presión, la isquemia por arteriopatía y la falta de sensibilidad por neuropatía periférica), se comentará algunas manifestaciones cutáneas típicas:
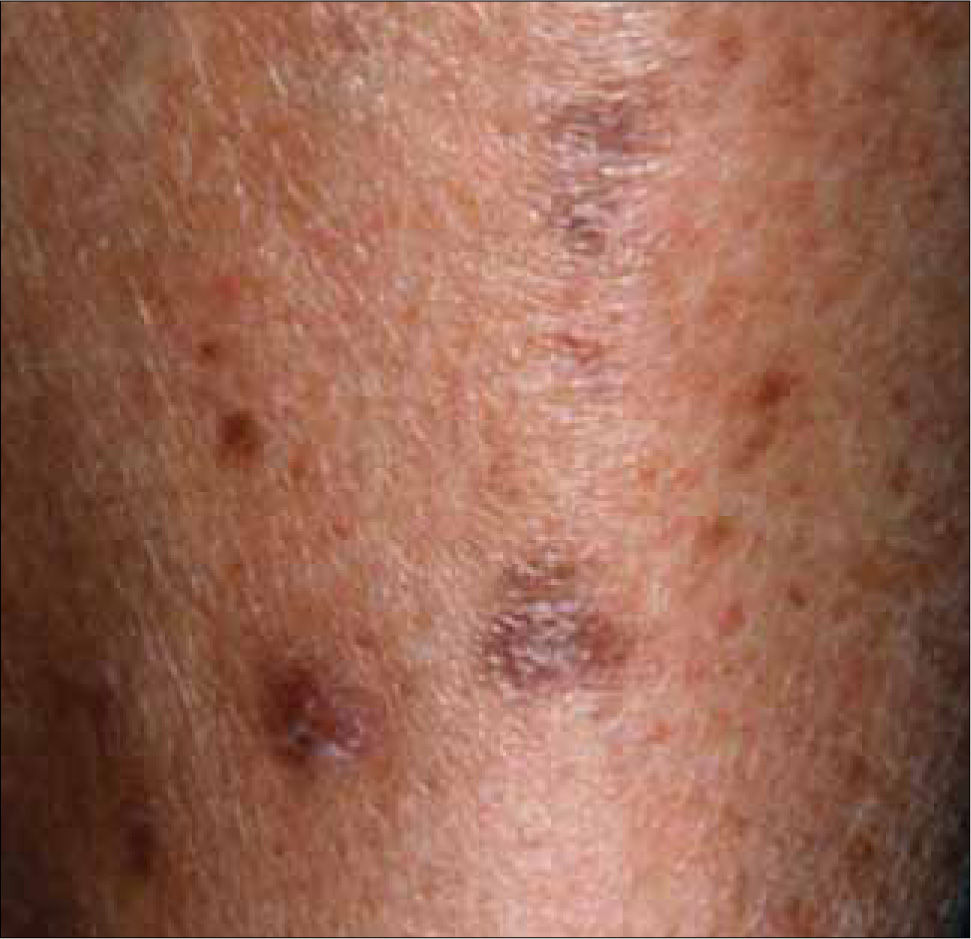
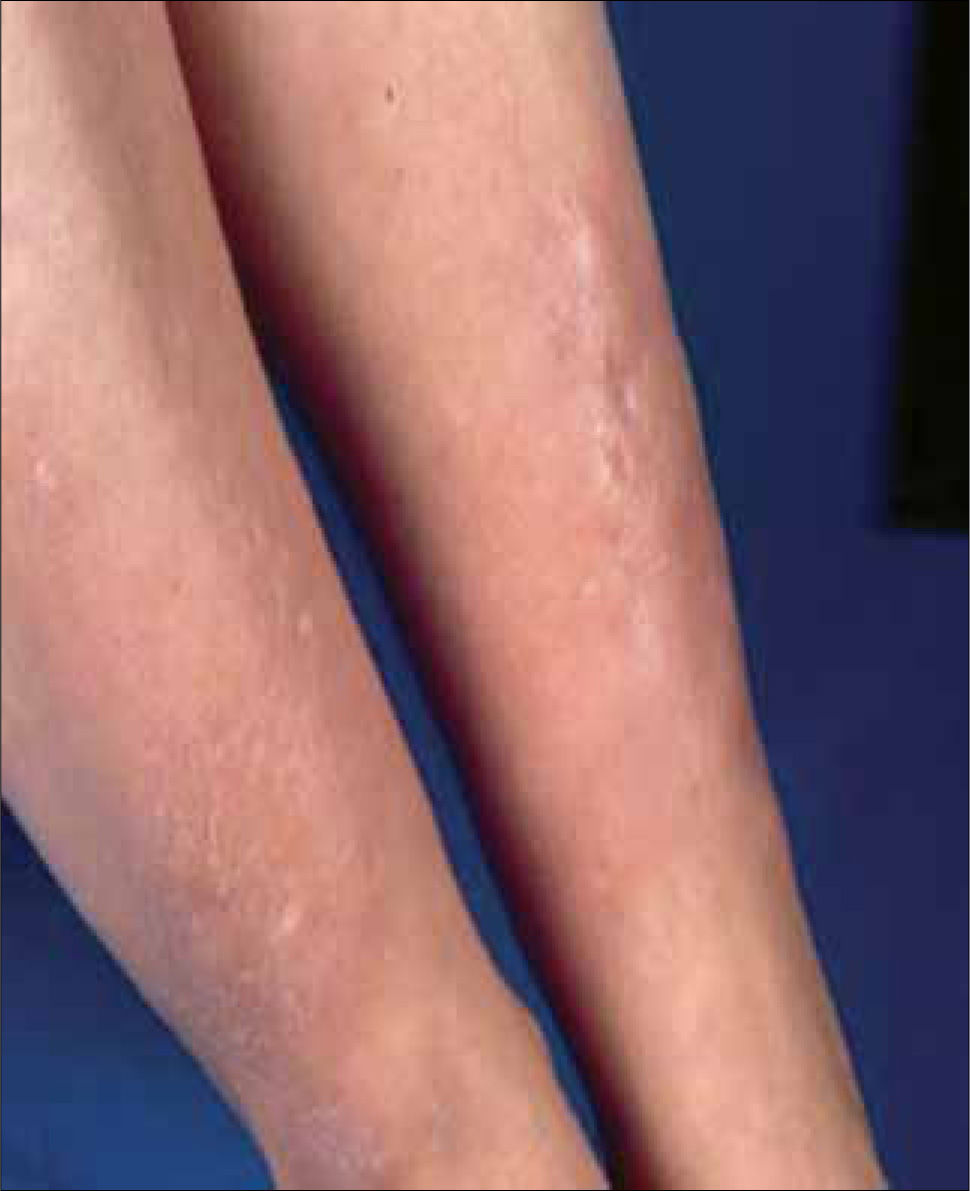
1. Dermopatía diabética: Son las lesiones más frecuentes en los diabéticos y se observan en el 50% de los pacientes. Se manifiestan como placas atróficas, de color rojizo o pardusco, irregulares y asintomáticas, que se localizan en la región pretibial. La causa es una microangiopatía. Generalmente, los pacientes con dermopatía diabética sufren otras complicaciones asociadas a la vasculopatía diabética, como retinopatía, neuropatía o nefropatía; por este motivo, es necesario descartar estas complicaciones. No existe ningún tratamiento eficaz. El trastorno cura por sí solo, dejando cicatrices deprimidas, atróficas e hiperpigmentadas. No existe ninguna correlación entre el control de la glicemia y el desarrollo de dermopatía diabética (Figura 2).

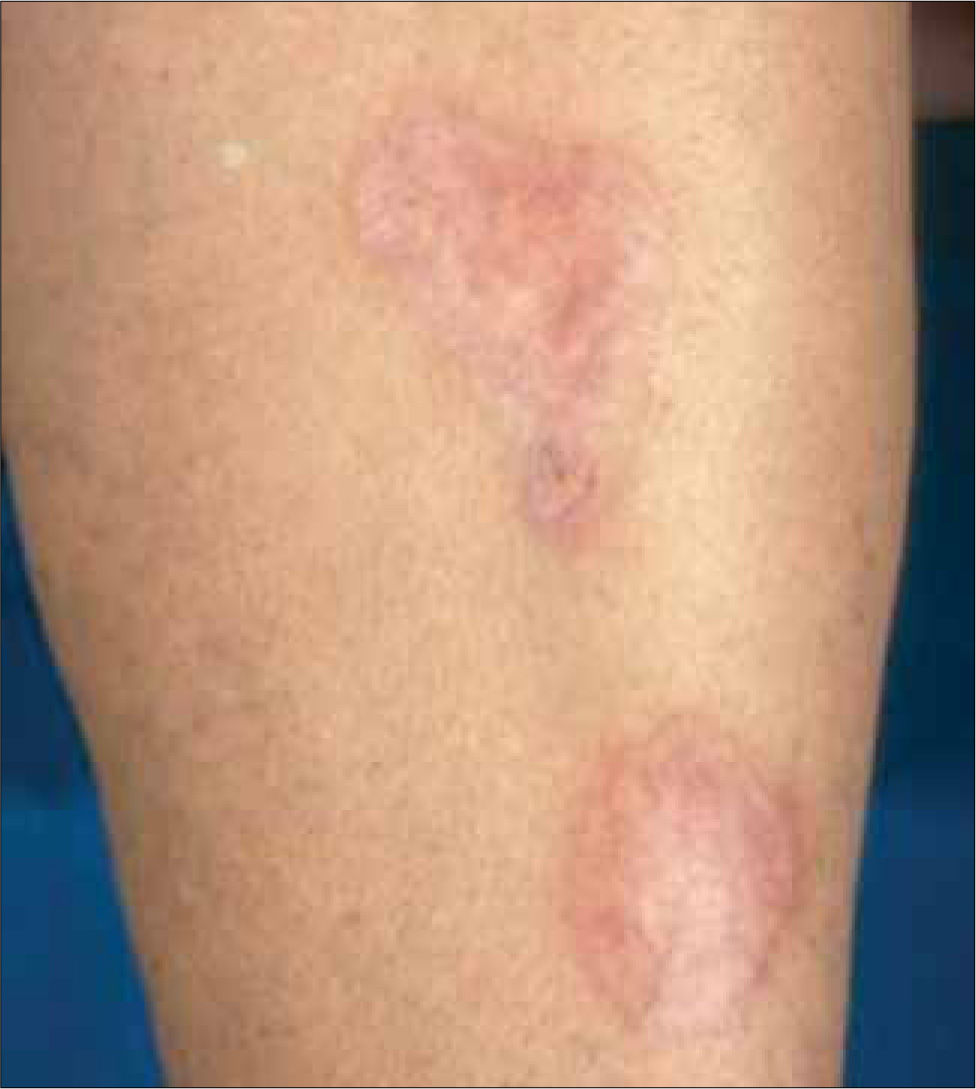
2. Necrobiosis lipoidea diabeticorum: Placas circunscritas atroficas pretibiales, de centro pardo amarillento, deprimido, que permite ver los vasos subyacentes y borde activo irregular, eritematoso, a veces sobreelevado. Las lesiones son indoloras y generalmente bilaterales. Entre el 66% y 75% de los pacientes con necrobiosis lipoídica tienen diabetes o intolerancia a la glucosa, pero menos del 1% de los diabéticos desarrolla esta patología. Se desconoce su etiopatogenia, aunque puede estar relacionada con la microangiopatía diabética. El tratamiento no resulta muy eficaz, y no es frecuente observar una remisión espontánea. Desgraciadamente un adecuado control de la glicemia no se acompaña de mejoría cutánea. Los corticoides tópicos e intralesionales pueden reducir la inflamación de las lesiones activas precoces y los bordes activos de las lesiones en crecimiento. Los apósitos de membranas semipermeables ayudan a tratar las lesiones ulceradas (Figura 3).

3. Acantosis Nigricans: Se manifiesta como engrosamiento aterciopelado y una hiperpigmentación de la piel, habitualmente en los pliegues. Es frecuente entre los pacientes con resistencia a insulina, y se asocia a la obesidad como parte del sindrome metabólico. En muchos casos, la reducción del peso permite invertir el proceso (Figura 4).

4. Escleredema diabeticorum: Este trastorno se caracteriza por una induración cutánea difusa, indolora, simétrica, no depresible, con eritema ocasional, que aparece fundamentalmente en la espalda. Es muy poco frecuente, suele afectar a diabéticos mayores de 40 años. Estos pacientes suelen necesitar insulina para el control de su enfermedad y sufren múltiples complicaciones. Es probable que el escleredema se deba a un depósito de glucosaminoglicanos en el tejido conjuntivo de la dermis. No existe ningún tratamiento efectivo, este trastorno suele pasar desapercibido.
5. Xantomas eruptivos: Se manifiestan como múltiples pápulas amarillentas o pardo rojizas que aparecen bruscamente. Al examen histológico se observa en las lesiones una infiltración de la dermis con macrófagos llenos de lípidos, que a diferencia de otros xantomas, representan triglicéridos. La causa consiste en una hipertrigliceridemia marcada. Las pápulas desaparecen al controlar la diabetes y las concentraciones de triglicéridos (17, 20, 21).
b) Manifestaciones cutáneas de los trastornos tiroideos: Las repercusiones de la actividad hormonal tiroidea sobre la piel son más visibles durante los estados de deficiencia o exceso que durante los procesos fisiológicos normales.
1. Hipotiroidismo: Puede ser un cuadro insidioso y se caracteriza por debilidad, fatiga, falta de concentración, constipación, aumento de peso, intolerancia al frío, disminución de la sudoración. La piel es seca rugosa o gruesa, fría y pálida, puede haber coloración amarillenta de palmas y plantas, puede haber ictiosis e hiperqueratosis palmoplantar. El pelo es seco, opaco, frágil, de crecimiento lento (por aumento del telógeno), puede haber alopecia difusa, del tercio externo de las cejas, región de la barba, vello axilar y pubiano. Existen casos de alopecia areata asociados a tiroiditis de Hashimoto. Las uñas son delgadas, frágiles, estriadas y de crecimiento lento.
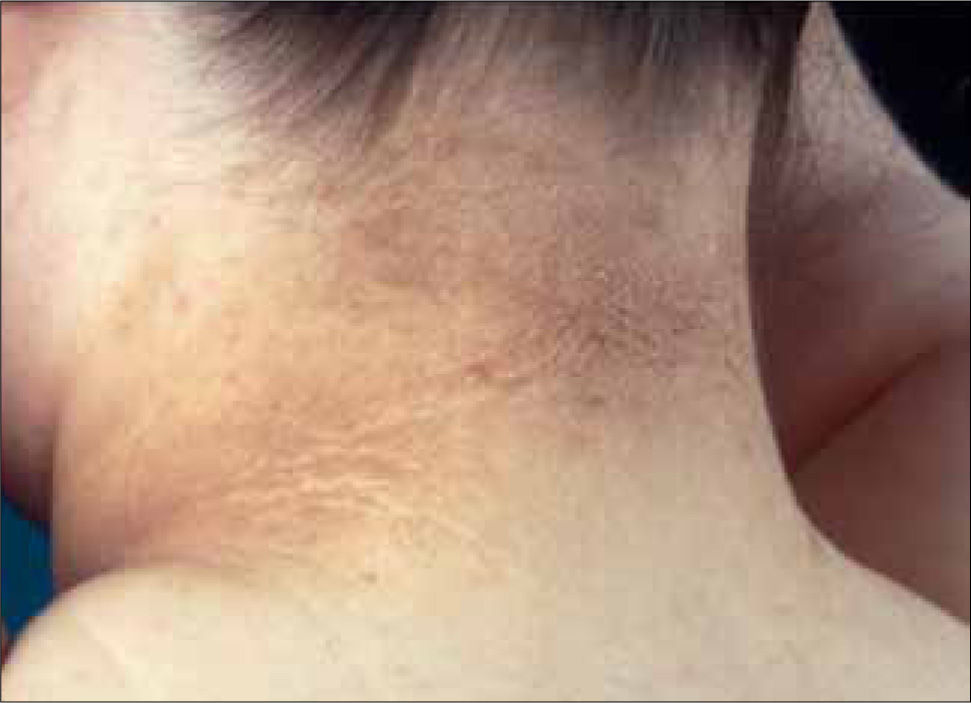
2. Hipertiroidismo: La piel se presenta fina, blanda, suave, aterciopelada, caliente y húmeda (por hiperhidrosis). Puede haber hiperpigmentación generalizada de tipo addisoniano o localizada (especialmente en cara, región periorbitaria, palmas y cicatrices). Por vasodilatación periférica puede aparecer flushing facial y eritema palmar. El pelo se ve fino, ralo, pueden haber zonas alopécicas. Los cambios ungueales se presentan en un 5% de los pacientes y consisten en uñas blandas, friables, con onicolisis o coiloniquia. Las llamadas "uñas de Plummer" son frecuentes de observar (onicolisis distal y contorno cóncavo). Entre la manifestaciones específicas del hipertiroidismo, encontramos el mixedema pretibial (Figura 5). Este se observa especialmente en el hipertiroidismo secundario a enfermedad de Basedow-Graves, aunque también puede verse en otras enfermedades tiroideas autoinmunes. Se asocia a exoftalmos y a acropaquia (dedos en "palillo de tambor"). Se manifiesta como placas edematosas con aspecto de piel de naranja, de color rosado o violáceo y duras al tacto, localizadas en la cara anterior del tercio inferior de las piernas. Puede haber hiperhidrosis e hipertricosis en la superficie. El estudio histopatológico revela un depósito de mucina en la dermis. La aparición del mixedema pretibial no guarda relación con la función tiroídea y puede desarrollarse en pacientes hiper, hipo o eutiroídeos. El tratamiento principal se realiza con corticoides tópicos potentes o bien, intralesionales. La plasmaféresis mejora transitoriamente y la cirugía no es recomendable (20, 22).
7Manifestaciones cutáneas asociadas a enfermedades del sistema nervioso central
Las enfermedades que afectan simultáneamente a la piel y al SNC son muy numerosas y de origen muy diverso. Las más conocidas son las que aparecen como resultado de trastornos del desarrollo embrionario, de origen hereditario o no hereditario, que afectan paralelamente a la piel y al SNC. Dichos trastornos, denominados también enfermedades neurocutáneas, se explican por el origen embrionario común de la piel y del SNC a partir del ectodermo embrionario.
Enfermedades neurocutáneasa) Neurofibromatosis tipo I (Enfermedad de von Recklinhausen):
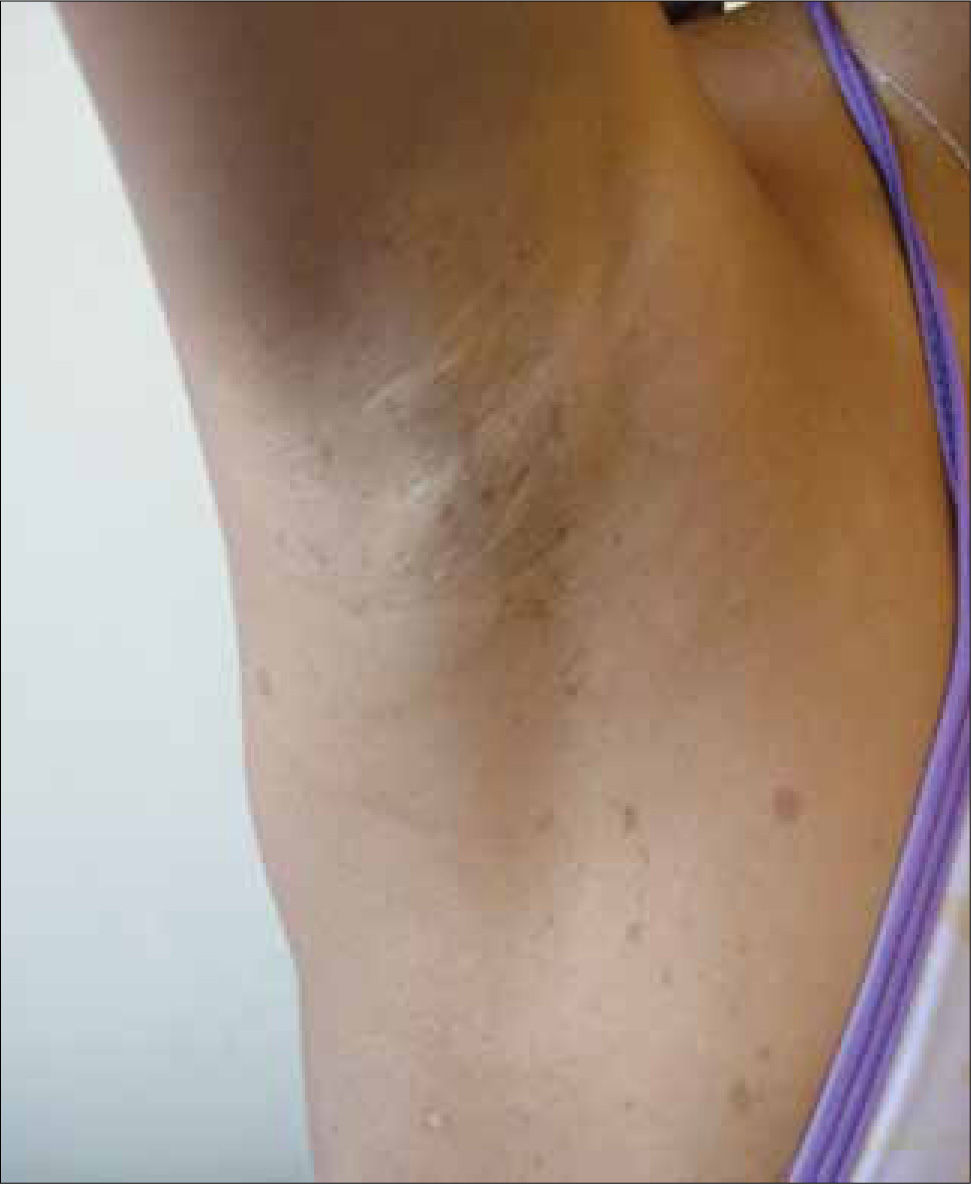
Enfermedad genética que se transmite de forma autosómica dominante y afecta a 1 de cada 3.000/RN. Existe gran diversidad de manifestaciones cutáneas y extracutáneas. En la piel produce manchas café con leche (presentes desde el nacimiento), pseudoefélides axilares y/o inguinales (signo de Crowe) (Figura 6) y neurofibromas (que aparecen al final de la infancia). A nivel sel SNC se desarrollan tumores (glioma óptico, astrocitomas y schwanomas), que pueden degenerar en el 5-10% de los casos. Son típicos los nódulos de Lish del iris presentes en más del 90% de los pacientes mayores de seis años Existen también lesiones óseas características como la displasia ala del esfenoides o el adelgazamiento de la cortical de los huesos largos, con o sin pseudoartrosis. El pronóstico es altamente variable. Alrededor del 20% de los pacientes desarrolla complicaciones durante la infancia (neurofibromas plexiformes, pseudoartrosis y escoliosis) y cerca del 15% tienen tumores viscerales o endocrinos. El 6% desarrolla lesiones malignas incluyendo tumores SNC. Requiere un enfoque multidisciplinario. Cuidadosa historia familiar, examen completo de piel y ojos. Exámenes radiológicos de huesos largos (23, 24). Figura 6)

b) Esclerosis tuberosa (Enfermedad de Bourneville): Síndrome neurocutáneo con herencia autosómica dominante con alto índice de mutaciones espontáneas. Se estima una incidencia de 1/10.000 RN. Generalmente el síndrome se manifiesta antes de los 5 años de vida, pero puede permanecer latente hasta la adolescencia.
Existe una gran variedad de manifestaciones cutáneas y extracutáneas, dentro de las principales características clínicas dermatológicas: Angiofibromas faciales (70-80%), placa fibrosa de la frente (20-40%), máculas hipomelanóticas lanceoladas (90-100%), placa de Zappa (40-50%), fibromas periungueales o tumores de Koenen (~ 80%), manchas café con leche. La afectación neurológica consiste en déficit mental, epilepsia, calcificaciones del SNC (hamatomas calcificados) y gliomas de la retina y del nervio óptico. También se observan quistes y angiolipomas renales y rabdomiomas cardíacos. Requiere enfoque multidisciplinario, examen neurológico, examen oftalmológico, EEG (si hay convulsiones), ECG, Ecocardiografía (si hay síntomas cardíacos), Ecografía renal, TC craneal, TC tórax (si hay síntomas pulmonares). Importante es el consejo genético a los padres (25).
c) Síndrome de Sturge-Weber: Asociación de nevus flameus o mancha “en vino de Oporto” facial, unilateral, en el territorio del la rama oftálmica del trigémino. Se asocia a angiomas leptomeningeos ipsilaterales que dan lugar a calcificaciones craneales responsables de convulsiones, hemiparesia, hemianopsia, con angiomatosis de la coroides y glaucoma. La mayoría de los casos son esporádicos. Cuando el nevus flameus se sitúa por debajo de la cisura palpebral, la afectación craneal es rara. La lesión es una malformación vascular que consiste en un número excesivo de capilares dilatados, bien delimitados a la dermis. Con los años la mancha en vino de Oporto aumenta la dilatación de las paredes de los vasos y aparecen dilataciones exofíticas similares a moras hacia la mitad de la etapa adulta. Puede haber hipertrofia gingival. Nuevas técnicas láser han mejorado el resultado estético del tratamiento. El láser de colorante pulsado es una alternativa específica para las lesiones vasculares. Puede lograrse un aclarado superior al 75% aunque es frecuente la recidiva con el paso del tiempo. Importante es la evaluación neurológica para evitar el daño cerebral (26-28).
d) Síndrome ataxia-telangiectasia: Trastorno autosómico recesivo causado por ataxia cerebelosa progresiva, telangiectasias oculares y cutáneas y una inmunodeficiencia variable. La inmunodeficiencia predispone al paciente a infecciones recurrentes pulmonares y de los senos paranasales, así como un aumento de neoplasia. Se considera una enfermedad radiosensible y tiene una incidencia de 1/40.000 nacimientos. Clínicamente hay coreoatetosis, nistagmus. Las telangiectasias aparecen entre los 2 y 8 años como vasos en forma de alambre en la conjuntiva bulbar y después en las zonas expuestas del pabellón auricular, cuello y pliegues flexores de las extremidades. Son frecuentes las alteraciones endocrinas asociadas. El 10% de los pacientes puede desarrollar antes de los 15 años algún cáncer, principalmente enfermedades linfoproliferativas. El tratamiento se limita a las infecciones, detección precoz del cáncer y consejo genético (29, 30).
e) Incontinencia Pigmenti (IP): Síndrome neurocutáneo autosómico dominante ligado al cromosoma X, predomina en mujeres (95%), alta letalidad masculina. Presenta lesiones cutáneas que siguen las líneas de Blaschko, reflejan mosaicismo. Clínicamente se presenta en 4 etapas evolutivas que se suceden o superponen, pueden desarrollarse in útero, y no siempre ocurren todas.
- Etapa I, inflamatoria/vesicular: En 90% pacientes en las primeras semanas o meses de vida. Vesículas y pústulas de distribución lineal siguiendo las líneas de Blaschko, extremidades y tronco. No afecta cara, sí cuero cabelludo.
- Etapa II, verrucosa: En 70% pacientes generalmente en los primeros meses de vida. Lesiones verrucosas y placas hiperqueratósicas de distribución lineal, habitualmente en las extremidades.
- Etapa III, hiperpigmentada: En 98% pacientes durante o poco después de la etapa anterior. Lesiones color café azulado de distribución lineal o espiral en tronco, extremidades, axilas o región pectoral. Aumentan hasta los 2 años.
- Etapa IV, hipopigmentada/atrófica: En 14-28% pacientes. Áreas hipopigmentadas más frecuentes en extremidades (77%). Se asocia alopecia cicatricial, agenesia de cejas y pestañas, onicodistrofia (50%). Lesiones cutáneas desaparecen hacia la adolescencia.
Dentro de las manifestaciones extracutáneas de la Incontinencia Pigmenti destacan: ausencia de pelo y glándulas sudoríparas, desarrollo asimétrico de las mamas, anomalías dentales (43%): manifestación no dermatológica más frecuente, distrofia ungueal, anomalías vasculares retinianas (30%), retraso mental, convulsiones. El diagnóstico diferencial incluye todas las enfermedades vesículo-bulosas del recién nacido. Las lesiones cutáneas no suelen requerir tratamiento. El tratamiento es sintomático. Debe realizarse una exploración oftalmológica básica a todos los lactantes, evaluación periódica del desarrollo dental y neurológico (31-33).
La autora declara no tener conflictos de interés, en relación a este artculo.