



Las malformaciones pulmonares congénitas son un grupo heterogéneo de alteraciones del desarrollo pulmonar que pueden producirse en distintas etapas de la embriogénesis, afectando al parénquima, la irrigación arterial, al drenaje venoso o ser una combinación de ellas. La nomenclatura y clasificación se ha modificado con frecuencia en el último tiempo, por lo que conocer la fisiopatología y etapa del desarrollo en que se producen es lo más relevante para su entendimiento.
El diagnóstico prenatal se hace en base a la ecografía y se apoya con resonancia magnética fetal, mientras que la radiografía y la TAC pulmonar son las principales herramientas para el diagnóstico posnatal. La presentación clínica es variable, desde la muerte por hidrops fetal, hasta pacientes que permanecen asintomáticos por años.
Existen alternativas terapéuticas prenatales para pacientes con alto riesgo de mortalidad intrauterina, y la extirpación posnatal es la conducta con más respaldo incluso en pacientes asintomáticos.
Congenital lung malformations are a heterogeneous group of disorders of lung development that may occur at different stages of embryogenesis, affecting the parenchyma, arterial supply, venous drainage or a combination of them. The nomenclature and classification has been changed frequently in recent times, so knowing the pathophysiology and stage of development in which they occur is the most relevant for understanding.
Prenatal diagnosis is based on ultrasound and fetal MRI, while pulmonary radiography and CT are the main test for post-natal diagnosis. The clinical presentation is variable, from fetal hydrops death, to patients who remain asymptomatic for years.
There are prenatal treatment options for patients at high risk of intrauterine mortality, and the postnatal removal is the behavior with more support, even in asymptomatic patients.
Las malformaciones pulmonares congénitas son un grupo diverso de trastornos del desarrollo y crecimiento broncopulmonar originados en las distintas etapas evolutivas del sistema respiratorio. Pueden afectar la vía aérea, el parénquima pulmonar, la irrigación arterial pulmonar, el drenaje venoso pulmonar, o una combinación de ellos1.
La incidencia reportada en la literatura es variable, con rangos entre 1:10.000 a 1:35.000 embarazos, sin embargo, algunos autores plantean que su incidencia real es mayor2,3.
Estos trastornos son consecuencia de alteraciones en la embriogénesis del pulmón y las vías respiratorias, y el tipo de lesión y su histología se relacionan directamente con el momento de la gestación y el nivel del árbol traqueobronquial en que se producen.
Para entender de mejor forma las distintas malformaciones pulmonares es importante conocer las etapas del desarrollo embrionario normal:
- 1.
Etapa embrionaria: ocurre durante las primeras 5 semanas de gestación.
A partir del intestino primitivo se desarrolla un divertículo ventral (respiratorio) desde donde inicia el desarrollo del tejido epitelial de todo el árbol respiratorio y de las vías aéreas mayores (destinada a convertirse en los 5 bronquios lobares).
Alteraciones en esta etapa embrionaria pueden manifestarse como algunas de las siguientes malformaciones pulmonares:
- -
Agenesia pulmonar, laríngea o traqueal
- -
Estenosis laríngea
- -
Traqueo-broncomalacia
- -
Malformaciones bronquiales
- -
Quistes pulmonares (como el quiste broncogénico)
- -
Fístula traqueoesofágica
- -
- 2.
Etapa pseudoglandular: entre la 5° y 16° semana de gestación.
En este período se produce la ramificación dicotómica de los bronquios que culmina con la formación de los bronquiolos terminales (preacinar). El tejido mesenquimático acompaña en su desarrollo a las vías aéreas y a partir de él desarrollará las estructuras cartilaginosas, musculares, linfáticas y sanguíneas.
Malformaciones que se originan en esta etapa son:
- -
Malformación congénita de la vía aérea pulmonar
- -
Linfangectasia pulmonar congénita
- -
Hipoplasia pulmonar
- -
Quistes pulmonares
- -
Secuestro pulmonar
- -
Hernia diafragmática congénita
- -
- 3.
Etapa canalicular: entre la 16° y 27° semana de gestación.
Formación de acinos y desarrollo de la irrigación sanguínea que se acerca a la vía aérea. Hacia el final de esta etapa aparece el surfactante y se hace posible la respiración con el aplanamiento progresivo del epitelio respiratorio.
Problemas en esta etapa pueden desencadenar en:
- -
Hipoplasia pulmonar
- -
Displasia acinar
- -
- 4.
Etapa sacular: desde la 28° a 36° semana de gestación.
Bronquiolos terminales se transforman en bronquiolos respiratorios y se forman racimos terminales llamados sáculos que permiten el intercambio gaseoso.
- 5.
Etapa alveolar: desde la semana 36 hasta los 2-3 años.
Aparecen septos secundarios y formación de estructuras alveolares que seguirán su desarrollo posteriormente.
Alteraciones en las etapas sacular y alveolar pueden tener como consecuencia el desarrollo de las siguientes patologías:
- -
Hipoplasia pulmonar
- -
Displasia acinar
- -
Displasia alvéolo-capilar
- -
Posterior al nacimiento y en forma progresiva hasta los 8 años, se va produciendo una maduración microvascular, con adelgazamiento de la pared alveolar y formación de la red capilar, una hiperplasia activa que aumenta el número de alvéolos y finalmente una hipertrofia alveolar que lleva a estas estructuras a su tamaño definitivo4–7.
CLASIFICACIÓN DE LA PATOLOGÍA PULMONAR CONGÉNITALa clasificación de las malformaciones congénitas pulmonares es controversial, pues existen diversos puntos de vista desde el cual pueden ser abordadas. Algunos autores utilizan la embriología como el factor principal para agrupar estas alteraciones según la etapa del desarrollo en que se producen8. Otros expertos categorizan las malformaciones de acuerdo a sus características morfológicas y radiológicas, dividiéndolas en aquellas que comprometen el pulmón en su totalidad y las que lo afectan parcialmente9,10. La clasificación más utilizada en la actualidad es la de Langston11, que desde el punto de vista fisiopatológico categoriza las malformaciones del tracto respiratorio en 5 tipos: Atresia bronquial, malformación pulmonar congénita de la vía aérea (antigua malformación adenomatoidea quística), secuestro broncopulmonar extralobar, hiperinsuflación lobar congénita y quiste broncogénico. Esta clasificación abarca aproximadamente al 90% de las alteraciones del desarrollo y crecimiento broncopulmonar que vemos en la práctica clínica, pero deja fuera otras anomalías congénitas menos frecuentes, como las malformaciones arteriovenosas.
Todas estas formas de clasificación tienen el inconveniente que, en la práctica, se combinan o sobreponen distintas categorías, por lo que un enfoque desde el tipo de lesión predominante en la malformación puede ser más claro y didáctico12,13. De esta forma, podemos tener tres grandes categorías: lesiones de predominio parenquimatoso, lesiones de predominio vascular o lesiones combinadas.
A continuación se describe la fisiopatología, evaluación y manejo de las patologías más relevantes y frecuentes que vemos en la práctica clínica.
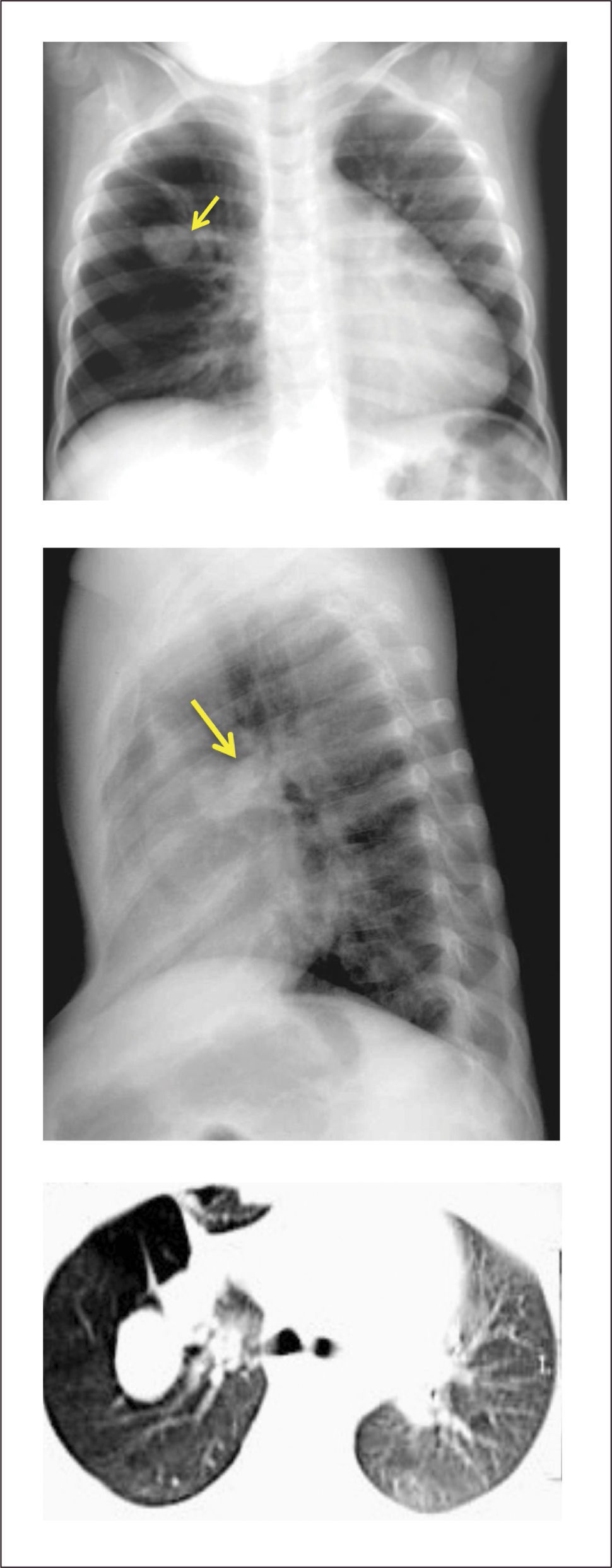
1Atresia bronquialLesión de predominio parenquimatoso que considera la atresia lobar, de bronquios segmentarios (la más frecuente) o de bronquios subsegmentarios. Sus causas no son claras, pero autores han propuesto como etiología más probable una injuria vascular producida en algún momento del desarrollo del árbol bronquial11,12.
Se describe asociada frecuentemente a secuestros broncopulmonares, y hasta en un 70% a la malformación congénita de la vía aérea pulmonar11,14,15, dependiendo de las diferencias de grado, nivel y momento del desarrollo en que se produjo la obstrucción bronquial.
Evaluación y diagnóstico. En muchas ocasiones la evolución es asintomática, por lo que el diagnóstico suele ser un hallazgo radiológico en niños mayores o en adultos11,16, sin embargo, la extensión de la ecografía prenatal y otras imágenes prenatales han aumentado su diagnóstico in útero14–17.
En recién nacidos, la zona distal al segmento atrésico puede atelectasiarse como resultado de la estasia mucosa. Esta imagen de zonas hipoventiladas puede confundirse con el pulmón poco aireado de recién nacidos con escaso clearence de líquido pulmonar en las primeras horas de vida, razón por la cual muchos autores recomiendan evitar una evaluación radiológica inmediata luego del parto16. Imágenes de tomografía axial computada y reconstrucciones 3D pueden también ser útiles en su diagnóstico16,18. (Figura 1).

Tratamiento. La resección quirúrgica está indicada en pacientes sintomáticos que presentan infecciones a repetición19,20. En aquellos asintomáticos la resección se programa en forma electiva, por el riesgo de infecciones y por la alta asociación a malformación congénita de la vía aérea pulmonar15,21.
2Malformación congénita de la vía aérea pulmonar (MCVAP)Antiguamente conocida como malformación adenomatoidea quística pulmonar, es una lesión de predominio parenquimatoso consistente en una masa multiquística benigna de tejido pulmonar no funcional, generalmente restringida a un lóbulo pulmonar, pero ocasionalmente más extendida (incluso bilateral), casi siempre con irrigación del territorio pulmonar. Se origina en un desarrollo embrionario pulmonar anormal, producido en la fase pseudoglandular, generando hiperplasia compensatoria de la vía aérea distal y reemplazo del parénquima normal por quistes de distintos tamaños y ubicación.
Fue descrita por primera vez por Chin et al. en 194922. La incidencia es de 1 por 25000 a 30000 embarazos25 y es la malformación pulmonar congénita más frecuente, llegando a un 25-30% de ellas24. Alrededor del 30% de los pacientes portadores desarrollan una falla respiratoria al nacer24.
La denominación actual fue propuesta por Stocker en el año 200223, abandonando el nombre de malformación adenomatoidea quística, debido a que en muchas ocasiones no se encontraban ni quistes ni malformaciones adenomatosas. De esta forma, agregó 2 categorías nuevas que se sumaron a los clásicos tipos I, II y III de 1977 que se basaba en el tamaño de los quistes y el probable punto de origen de desarrollo de la malformación a lo largo de la vía aérea25:
- -
Tipo 0: se origina a partir de la tráquea o tejido bronquial y tiene una baja incidencia (1-3%). Los quistes son de hasta 0.5cm de diámetro y afectan a todos los lóbulos pulmonares. En general son incompatibles con la vida y se asocian con anomalías cardíacas graves.
- -
Tipo I: quiste único grande o múltiples mayores a 2cm de diámetro. Es el más frecuente (50-70%) y el con mejor pronóstico. La malformación ejerce un efecto de masa que comprime el pulmón sano adyacente y puede generar dificultad respiratoria en el recién nacido.
- -
Tipo II: múltiples quistes de menos de 2cm que afectan a 1 lóbulo pulmonar y tiene origen bronquiolar. Es el segundo tipo más frecuente (20-40%) y tiene una alta asociación con secuestro pulmonar y malformaciones de otros sistemas (cardíacas, hernia diafragmática, atresia de esófago, agenesia renal)27.
- -
Tipo III: masa sólida de origen bronquiolar/alveolar que puede desplazar y comprimir el pulmón contralateral y las estructuras mediastínicas, provocando hipertensión pulmonar e hidrops fetal24. Equivale aproximadamente al 10% de las MCVAP.
- -
Tipo IV: tiene un origen acinar distal con grandes quistes periféricos de hasta 7cm de diámetro, y corresponde a un 10 a 15% de las MCVAP. Se ha propuesto como una etapa evolutiva del blastoma pleuropulmonar tipo 111,28.
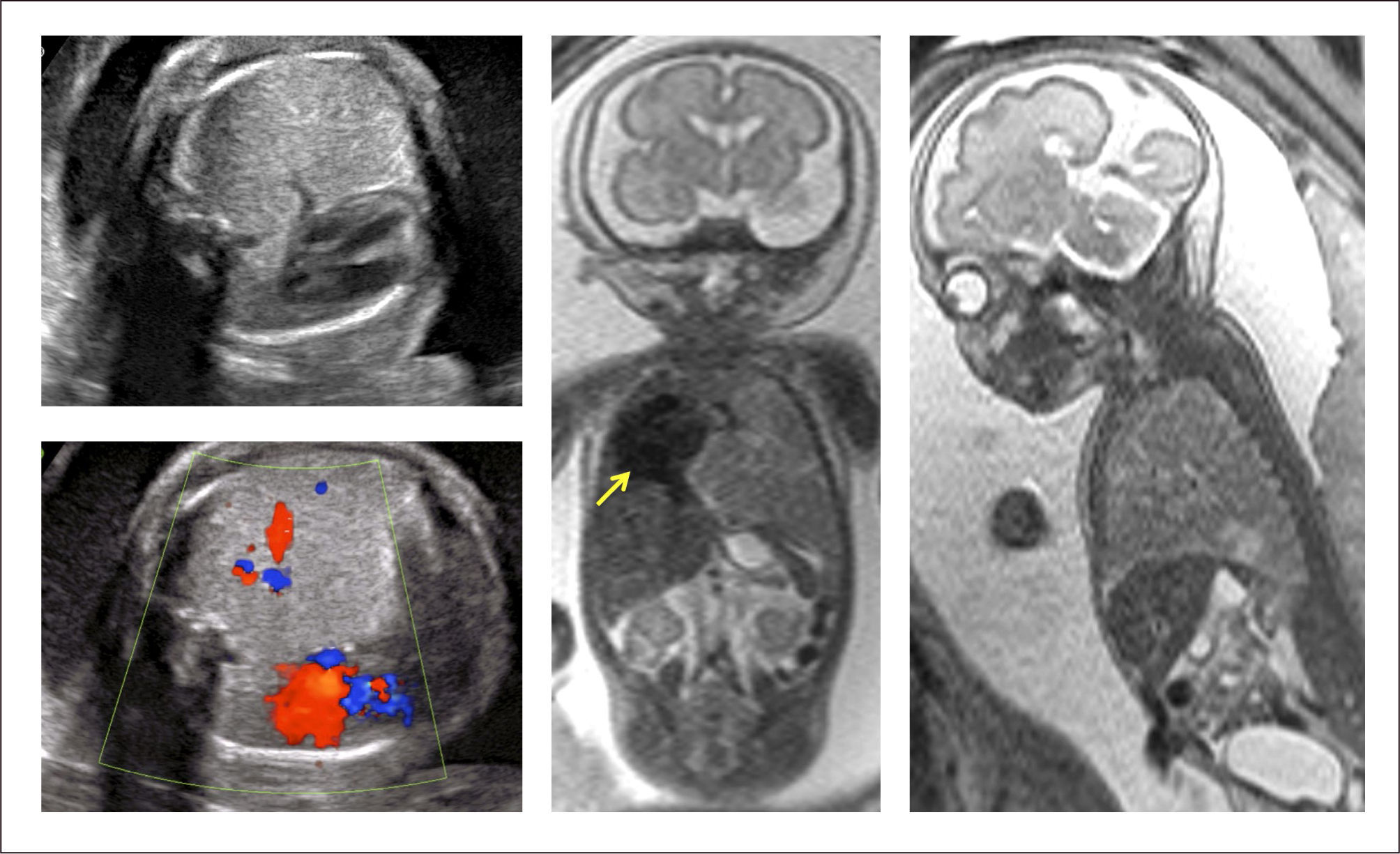
Evaluación y diagnóstico. Existe un amplio espectro de manifestaciones clínicas, que pueden ir desde la muerte fetal (por hidropesía o hipoplasia pulmonar) hasta la falta absoluta de síntomas24,29. La clínica más frecuente son las infecciones pulmonares recurrentes, dificultad respiratoria, neumotórax, hemotórax y hemoptisis, pero se describe que hasta un 90% de los portadores puede permanecer asintomático en el período neonatal33.
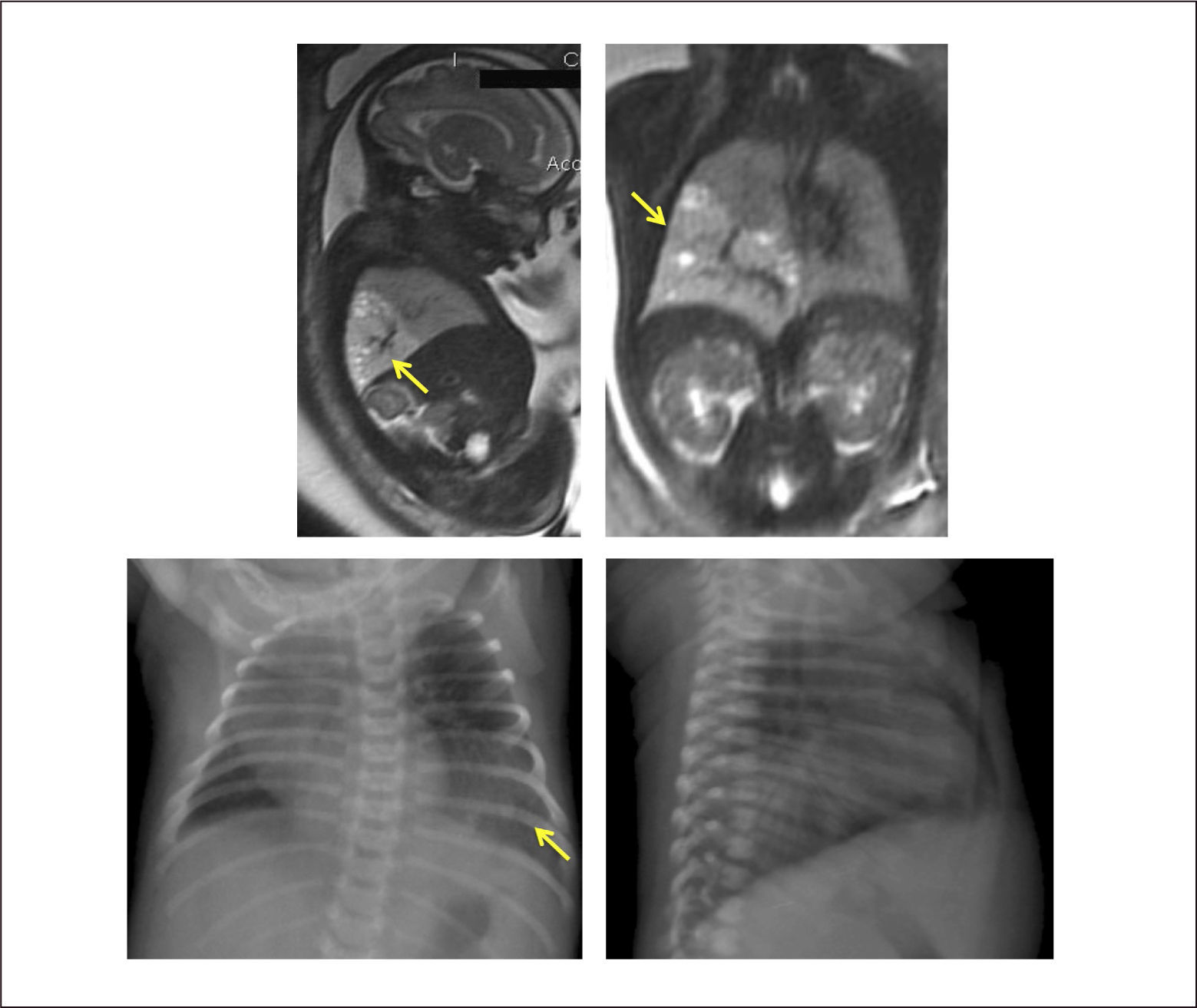
La extensión de la evaluación ecográfica prenatal ha permitido que este tipo de lesiones se detecten cada vez con más frecuencia antes del nacimiento, pudiendo identificar desde una zona quística en el tórax, hasta una hidrops fetal, que puede desarrollarse hasta en el 40% de los casos, con alto riesgo de mortalidad35. La resonancia magnética fetal ayuda en el diagnóstico diferencial (hernia diafragmática, duplicación intestinal anterior, secuestro pulmonar, enfisema lobar congénito) y en identificación de anomalías asociadas. (Figura 2).

El estudio en el período neonatal incluye la radiografía de tórax, que puede mostrar múltiples quistes de paredes finas, con ocupación interior o contenido aéreo, y desplazamiento de estructuras mediastínicas. La tomografía permite determinar el tamaño de los quistes, la extensión anatómica y la identificación de otras alteraciones asociadas no visualizadas con Rx o ecografía25,28. (Figura 3).

Tratamiento. La literatura describe alrededor de un 20% de resolución espontánea de este tipo de lesiones durante la gestación, pero para aquellas asociadas a hidrops, con riesgo de mortalidad cercano al 100%, existen intervenciones fetales que han demostrado reducción de ese riesgo:
- -
Corticoides prenatales: pacientes con MCVAP de alto riesgo de predominio sólido-microquístico, sometidos a inyecciones prenatales maternas de betametasona, han evidenciado reducción del volumen de la lesión, con resolución del hidrops cercano al 80% y sobrevida en torno al 85%34,36.
- -
Shunt toracoamniótico: pacientes portadores de lesiones de alto riesgo con múltiples comunicaciones y predominio macroquístico, se benefician de la instalación de una comunicación entre los grandes quistes y la cavidad amniótica que permite su drenaje. Los reportes describen que la técnica permite una reducción del volumen de la masa entre un 50 y un 80%, con sobrevidas cercanas al 75%37.
- -
Cirugía fetal: la lobectomía, neumonectomía o la resección de la masa en fetos menores de 30 semanas que no respondieron a los corticoides prenatales es una alternativa terapéutica para aquellos fetos portadores de lesiones de predominio sólido o microquístico con alto riesgo de mortalidad. La literatura describe distintas aproximaciones técnicas, todas las cuales buscan reducir la compresión de las estructuras y el hidrops fetal38,39.
- -
Cirugía posnatal: cuando el RN evoluciona sintomático la indicación quirúrgica es inmediata, y la resección de la malformación, generalmente por lobectomía, es el tratamiento de elección. La evidencia actual sugiere que debe resecarse aun siendo asintomático, debido al riesgo de infecciones, neumotórax y de malignización futura40. Técnicas por toracotomía o toracoscopía son descritas, evidenciando menores complicaciones postoperatorias con las técnicas mínimamente invasivas41–43.
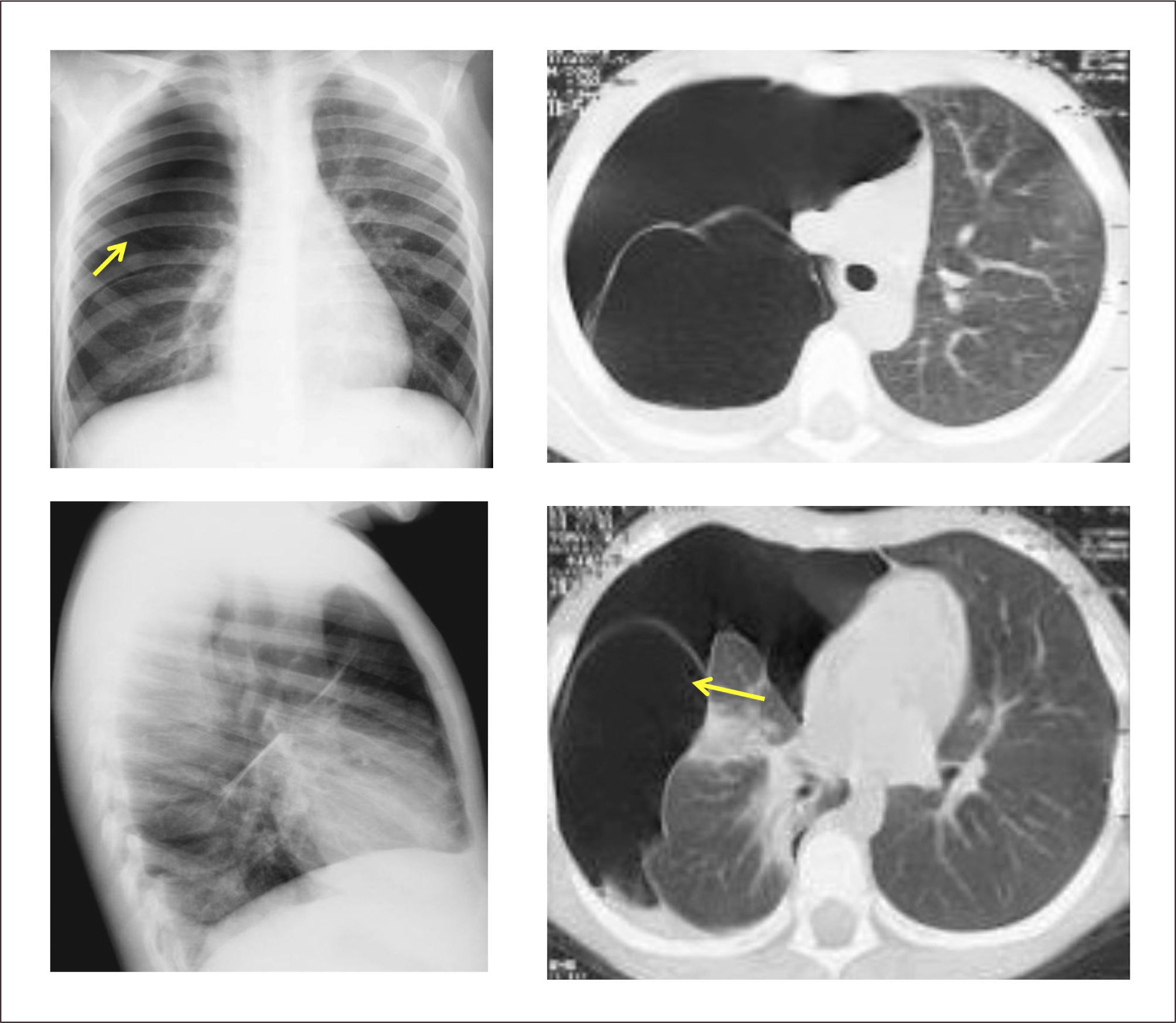
Lesión de predominio parenquimatoso cuya incidencia se estima entre 1:20000 a 1:30000 nacimientos. Consiste en una hiperinsuflación de uno o más lóbulos pulmonares que produce compresión del parénquima restante y desplazamiento mediastínico. Su origen se encuentra en un estrechamiento bronquial intrínseco o extrínseco que lleva a atrapamiento aéreo. Las causas principales son la alteración del desarrollo del cartílago bronquial y la estenosis bronquial idiopática, las que explicarían el 50% de los casos, siendo desconocida la causa de la restante mitad25,30,44.
Evaluación y diagnóstico. 1/3 de los pacientes presenta síntomas en el período de recién nacido y un 50% después de los 6 meses de vida, y un bajo porcentaje puede mantenerse asintomático por varios años. La sintomatología respiratoria es variable y dependiente de la distensión progresiva del lóbulo comprometido, pudiendo presentar dificultad respiratoria leve inicial y llegar a una falla respiratoria aguda con el paso de los días44,45.
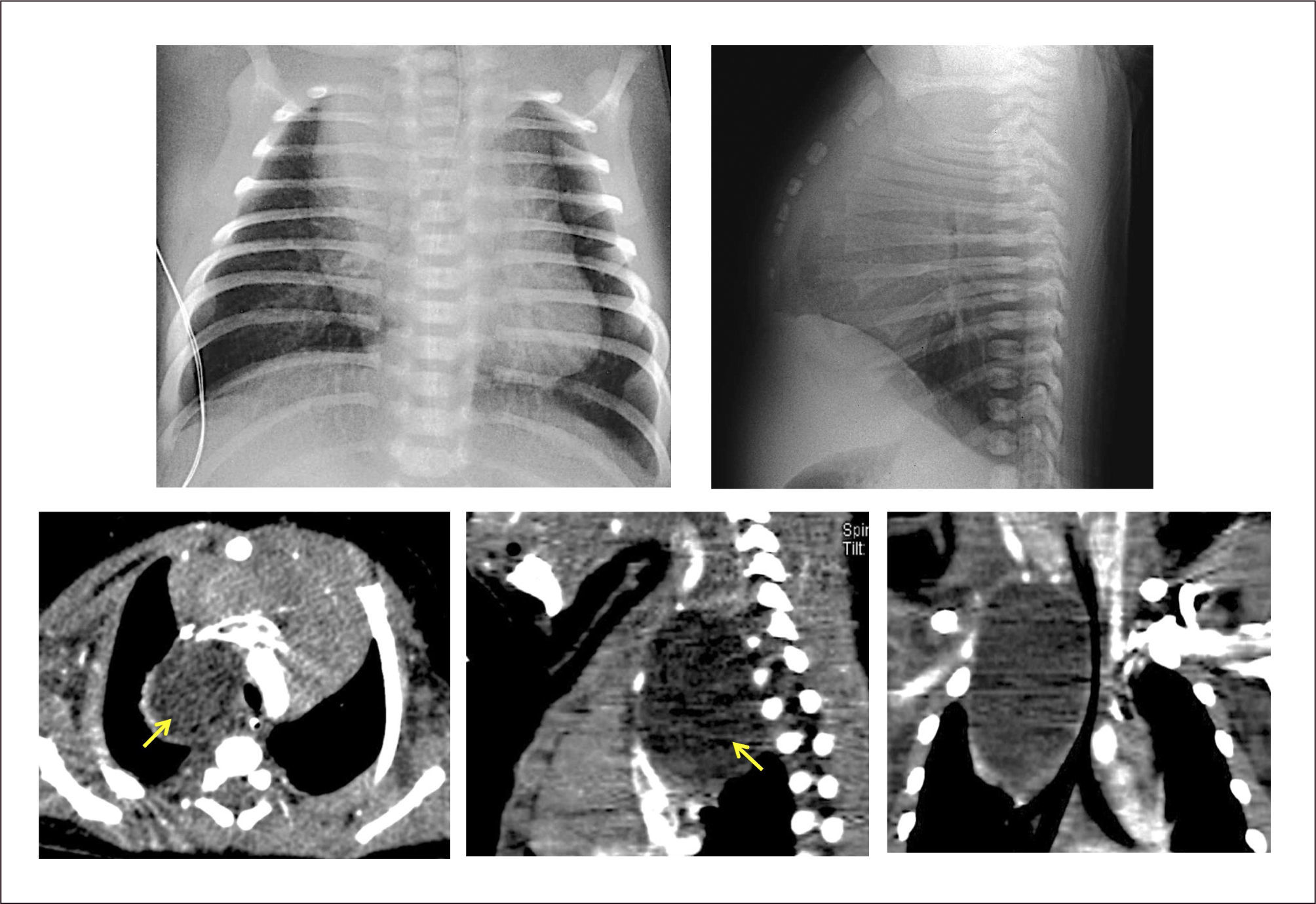
La ecografía prenatal puede identificar una lesión hiperecogénica homogénea, difícil de distinguir de otras malformaciones pulmonares, pero la gran mayoría se diagnostica posterior al nacimiento en base a un paciente con dificultad respiratoria a quien se realiza una radiografía de tórax que demuestra hiperinsuflación y menor trama pulmonar de un lóbulo, desplazamiento del mediastino y zonas atelectásicas en las zonas adyacentes por compresión30. La TAC pulmonar es importante para demostrar la presencia de vasos pulmonares atenuados en el lóbulo hiperinsuflado, la zona de obstrucción bronquial y los límites de la lesión, lo que permite diferenciarlo de un neumotórax u otras alteraciones30,46. (Figura 4).

Tratamiento. La lobectomía por toracotomía o toracoscopía es el tratamiento de elección para los pacientes sintomáticos, programándose de manera electiva en aquellos con síntomas mínimos o asintomáticos47. La evolución postquirúrgica es en general favorable y no presenta mayores secuelas y la pérdida de volumen pulmonar es compensada progresivamente con el sobrecrecimiento del tejido remanente, llegando a tener una función normal a largo plazo44,46.
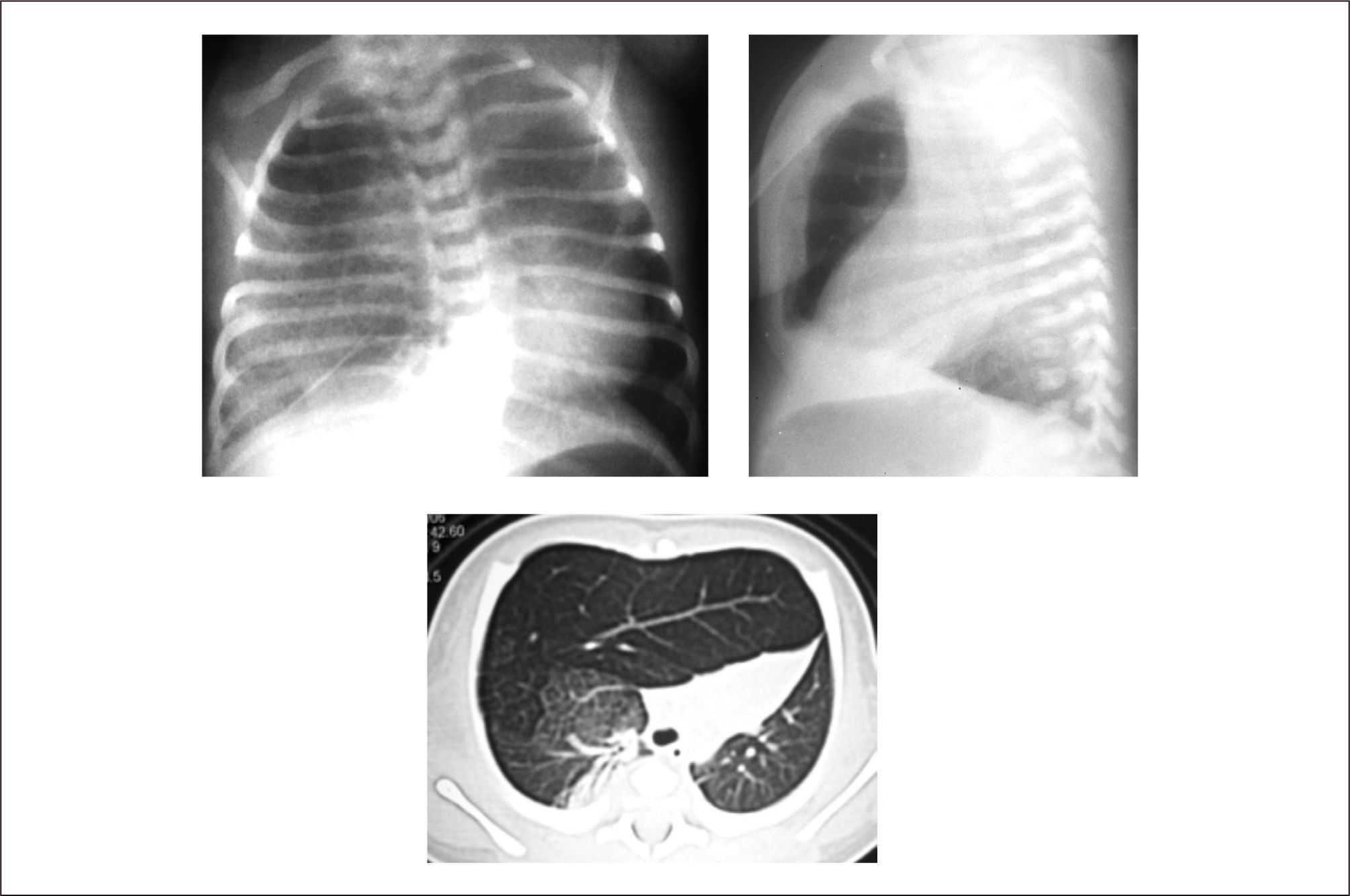
4Quiste broncogénicoLesión de predominio parenquimatoso que surge a partir de la gemación aberrante del divertículo traqueal del intestino anterior en desarrollo, que resulta en una ramificación traqueobronquial anormal. Clásicamente se trata de una formación quística única, de contenido líquido o mucoso y habitualmente no comunicada con la vía aérea. Su ubicación más frecuente es a nivel mediastínico (cercano a la carina), pero pueden encontrarse en medio de cualquier zona de parénquima pulmonar o incluso subpleurales. Pueden asociarse a otras malformaciones como el enfisema lobar congénito o la MCVAP19,20.
Evaluación y diagnóstico. En la mayor parte de los recién nacidos portadores no existen síntomas, pudiendo aparecer posteriormente síntomas de compresión de la vía aérea o infecciones recurrentes cuando presentan comunicación con la vía aérea48.
El diagnóstico suele ser un hallazgo en una radiografía de tórax en la que se identifica una imagen redondeada de contenido sólido (o aéreo si tiene comunicación con la vía aérea) especialmente a nivel de mediastino medio, pudiendo además evidenciarse desplazamiento de sus estructuras.
La TAC pulmonar o la resonancia magnética pueden confirmar la naturaleza quística o sólida de la masa y ayudar en el diagnóstico diferencial de neumatoceles, neumonía abscedada o quiste hidatídico20, (Figura 5).

Tratamiento. La resección quirúrgica del quiste por toracotomía amplia o videotoracoscopía es el tratamiento de elección, o eventualmente la lobectomía, si el quiste es intrapulmonar. Una vez extirpado, permite una buena expansión del parénquima pulmonar adyacente y la recuperación completa de la función pulmonar. También se describe como procedimiento temporal o paliativo la aspiración mediastinal, transparietal o transbronquial del quiste cuando los pacientes tienen alguna contraindicación para la cirugía48.
5Agenesia, aplasia e hipoplasia pulmonarLesiones de predominio vascular caracterizadas por una falla en el desarrollo pulmonar de distintos grados, cuya etiología no es conocida, pero existen factores genéticos y teratogénicos a los que se han descrito asociados. Con frecuencia se presentan junto a malformaciones cardíacas, esqueléticas, gastrointestinales, vasculares o genitourinarias19,49.
- -
Agenesia pulmonar: ausencia de pulmón, bronquios y arteria pulmonar uni o bilateral. Las bilaterales son incompatibles con la vida, y la unilateral izquierda se asocia a peor pronóstico (50% mortalidad en período neonatal).
- -
Aplasia pulmonar: presencia de un bronquio rudimentario que termina en un fondo de saco, con ausencia de tejido pulmonar y arteria pulmonar.
- -
Hipoplasia pulmonar: presencia de un árbol bronquial y arterial hipoplásicos, con disminución variable del tejido pulmonar19,48. La hipertensión pulmonar (HP) asociada a la hipoplasia vascular, es el principal marcador de las posibilidades de sobrevida en recién nacidos12.
Evaluación y diagnóstico. La clínica dependerá del grado de alteración del desarrollo pulmonar y de la asociación a otras malformaciones, pero puede ir desde RN asintomáticos hasta aquellos que presentan una dificultad respiratoria severa asociada a HP que los lleve a la muerte.
En pacientes asintomáticos se puede una auscultación asimétrica y en la Rx de tórax encontrar la falta de aireación del pulmón afectado, desviación de las estructuras torácicas hacia el mismo lado e hiperinsuflación compensatoria del pulmón contralateral y herniación a través de la línea media. La angio TAC de tórax puede demostrar la ausencia total o parcial de la arteria pulmonar y del parénquima y la broncoscopía la ausencia o reducción de calibre del bronquio del lado afectado.
Tratamiento. El manejo de RN con hipodesarrollo pulmonar primario en general es conservador y depende fundamentalmente del grado y severidad de la alteración, pero también de las malformaciones asociadas50. En caso de que se trate de una hipoplasia pulmonar secundaria, la corrección quirúrgica de la causa primaria es parte importante del manejo (por ejemplo, hernia diafragmática congénita), realizándose de manera electiva cuando se ha logrado la estabilidad hemodinámica del paciente.
6Síndrome de la CimitarraTambién conocido como síndrome del pulmón hipogenético, es una lesión combinada vascular y parenquimatosa, caracterizada por una conexión anómala, parcial o total, del drenaje venoso pulmonar derecho que puede desembocar en la vena cava inferior, unión cavoatrial o aurícula derecha, y que característicamente se asocia hipoplasia del pulmón derecho y dextrocardia, aunque también puede asociarse a otras alteraciones cardiovasculares (25% de los pacientes presenta malformaciones cardíacas asociadas)51,52.
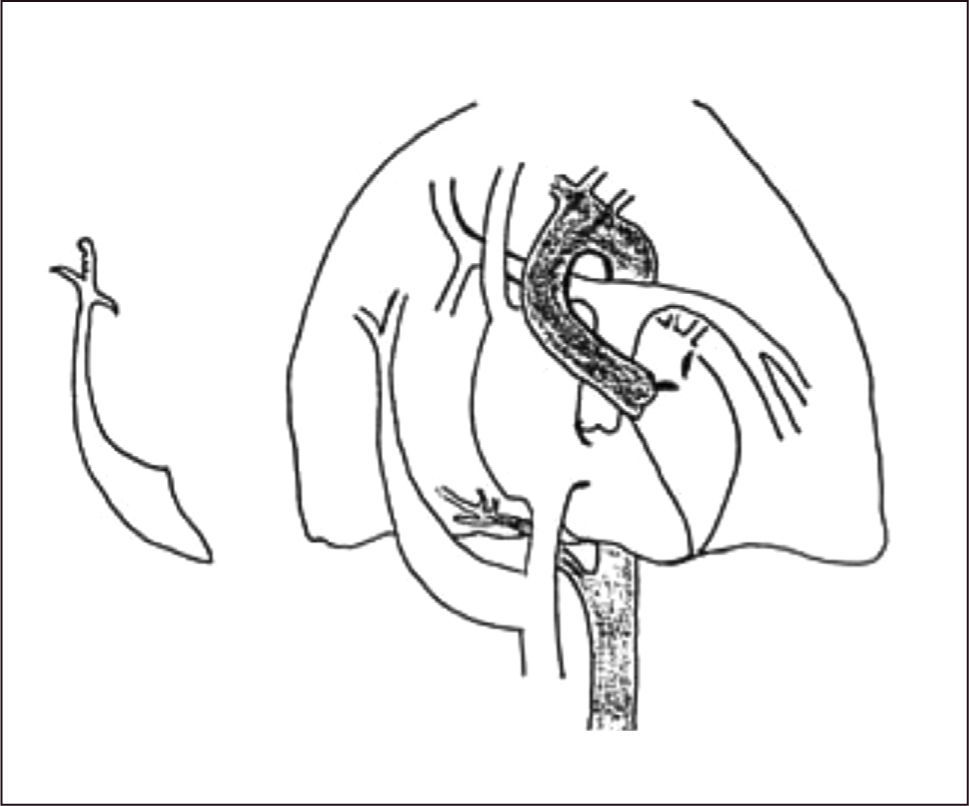
Su nombre se debe a que el vaso aberrante se visualiza radiológicamente con la forma de una “cimitarra”, sable de hoja curva característico del medio oriente.
Evaluación y diagnóstico. Los hallazgos clínicos son variables, pero los pacientes pueden mantenerse asintomáticos hasta la edad adulta, o debutar precozmente con signos de falla cardíaca congestiva por hiperflujo pulmonar, que con el tiempo lleva al paciente a una hipertensión pulmonar. Niños mayores también pueden presentar infecciones pulmonares recurrentes19,20.
La radiografía anteroposterior puede demostrar el signo de la cimitarra (Figura 6), característica opacidad vertical con esta forma en la mitad inferior del hemitórax derecho, asociado a una hipoplasia del mismo pulmón. La TAC pulmonar y la RNM pueden confirmar el diagnóstico y la presencia de otras malformaciones asociadas, identificándose la reconstrucción 3D como una buena herramienta para determinar el recorrido completo del vaso, previo a la corrección quirúrgica20,53.
: 187-189.")
Tratamiento. En pacientes sintomáticos se realiza la corrección quirúrgica del drenaje pulmonar derecho anómalo a la aurícula izquierda54. En pacientes con neumonía recurrente asociada, puede ser necesaria la pneumonectomía total o parcial.
7Secuestro pulmonarLesión vascular y parenquimatosa caracterizada por la presencia de tejido pulmonar no funcionante, sin comunicación con el árbol bronquial y que recibe irrigación de la circulación sistémica, generalmente desde la aorta. Corresponde aproximadamente al 6% de todas las malformaciones pulmonares congénitas y su etiología no es conocida.
Clásicamente se clasifica en secuestros extralobares (SE) e intralobares (SI), siendo 3 veces más frecuente el secuestro pulmonar intralobar.
El SE es característicamente una masa de tejido pulmonar anormal separada del parénquima normal por un revestimiento pleural propio. Recibe irrigación desde la aorta torácica o abdominal y el retorno venoso es hacia el sistema ácigos o venas cavas, en la mayoría de los casos11,57. Generalmente se presenta aislado, pero ocasionalmente se puede asociar a defectos cardíacos, hipoplasia pulmonar, anomalías vertebrales y a hernia diafragmática11,16,20,55; también se presenta asociado a MCVAP tipo II16,56.
El SI presenta tejido pulmonar anómalo integrado al parénquima pulmonar normal, con el que comparte revestimiento visceral y pleural. La irrigación proviene de la aorta y el drenaje venoso ocurre hacia las venas pulmonares o a un colector venoso. Clásicamente se ubica en los lóbulos inferiores, especialmente el izquierdo20.
Evaluación y diagnóstico. Más de la mitad de los SI son asintomáticos y aparecen como un hallazgo radiológico en la adolescencia. Los pacientes sintomáticos pueden desarrollar infecciones recurrentes, tos crónica o hemoptisis.
En el caso del SE, la mayoría de los casos son detectados en la infancia, siendo 4 veces más frecuente en el género masculino. La sintomatología más frecuente es la tos crónica y neumonías a repetición25,57.
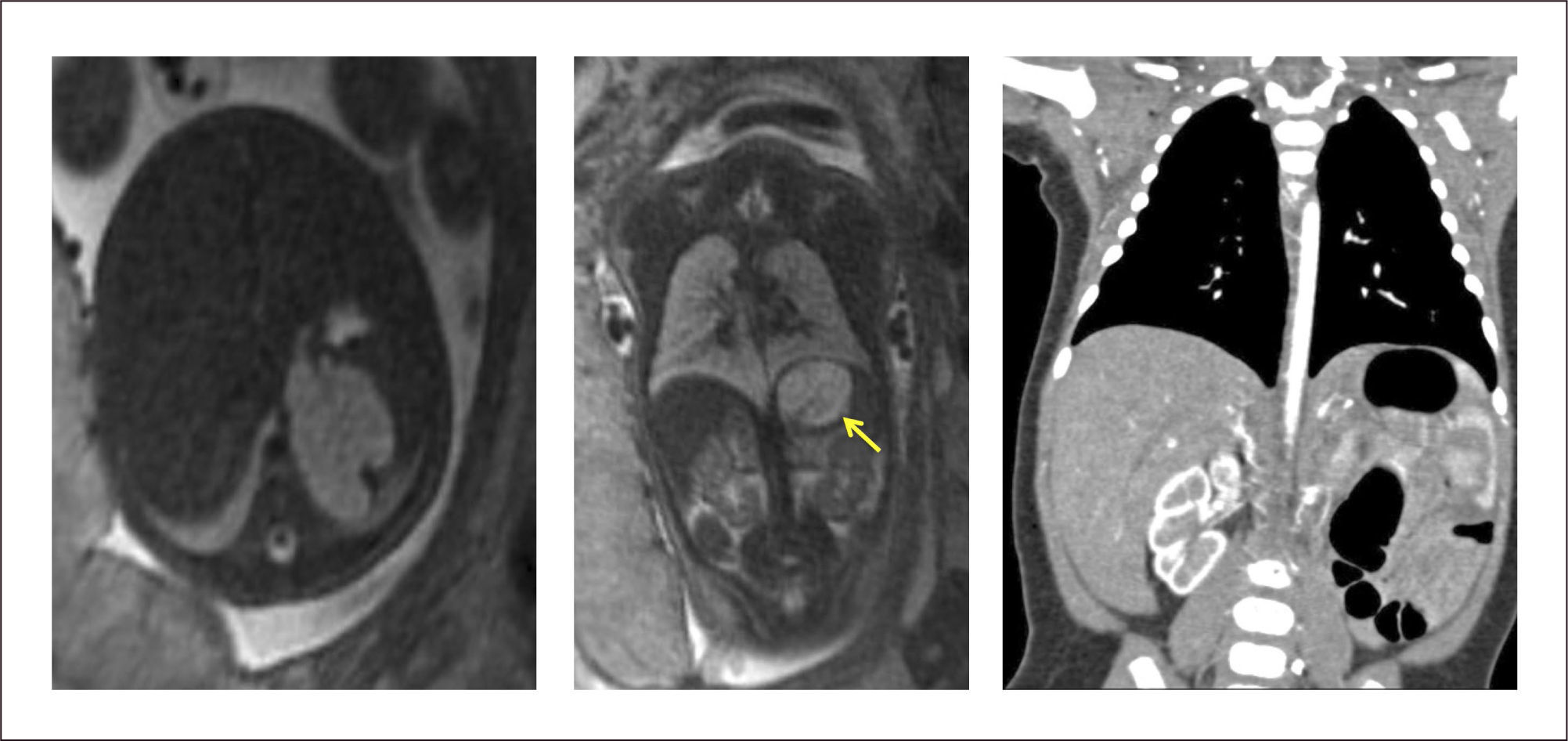
La evaluación ecográfica prenatal puede identificar una masa hiperecogénica, pero con el paso de las semanas se puede confundir con el tejido pulmonar normal adyacente que va aumentando su ecogenicidad hacia el final de la gestación. La resonancia magnética ayuda en el diagnóstico diferencial. (Figura 7).

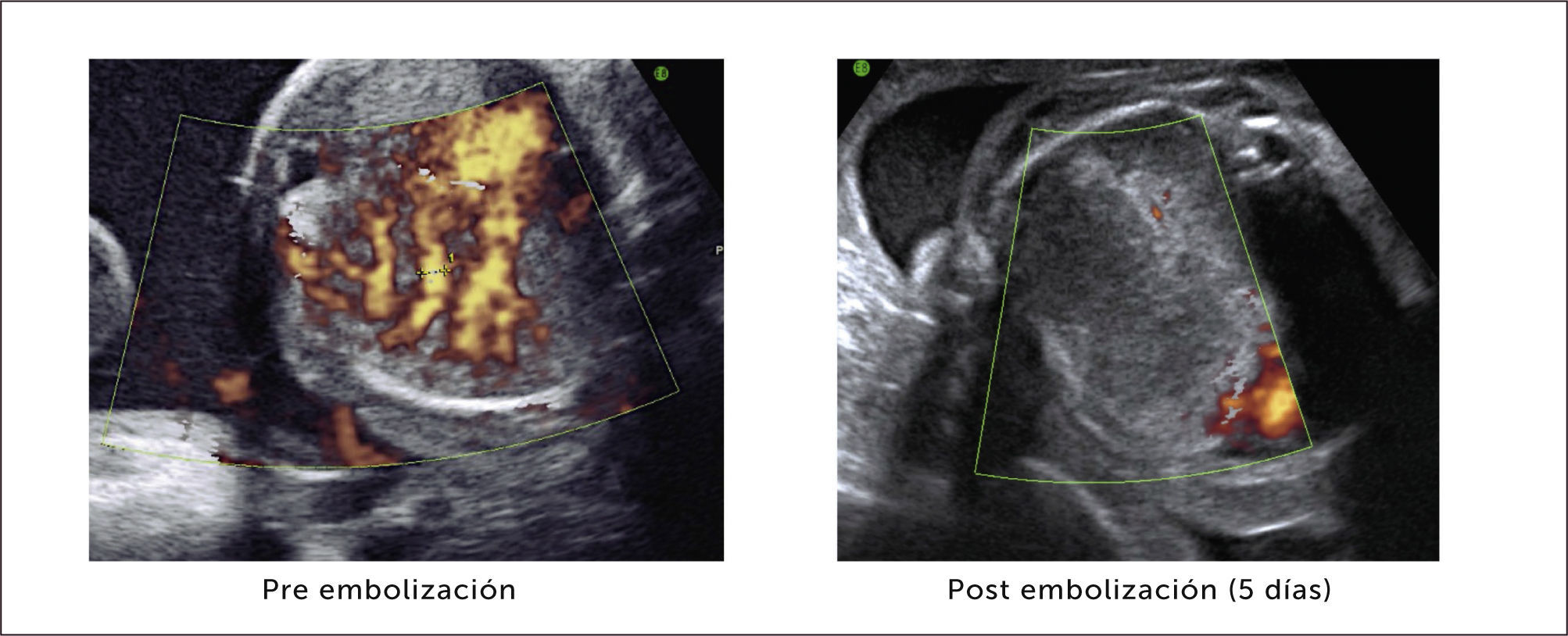
La evaluación posnatal con Rx de tórax, demuestra típicamente una consolidación en una de las bases (generalmente la izquierda), siendo la angio TAC la que permite identificar el vaso sistémico anómalo y eventuales malformaciones asociadas. (Figura 8).

Tratamiento. La resolución en ambos tipos de secuestro es siempre quirúrgica, incluso en los casos asintomáticos por el riesgo de complicaciones posteriores, realizando una segmentomía o lobectomía a cielo abierto o toracoscópica48,58,59. Algunos grupos consideran la embolización arterial como una alternativa que permite la reducción del tamaño de la lesión, para una eventual extirpación posterior60,61.
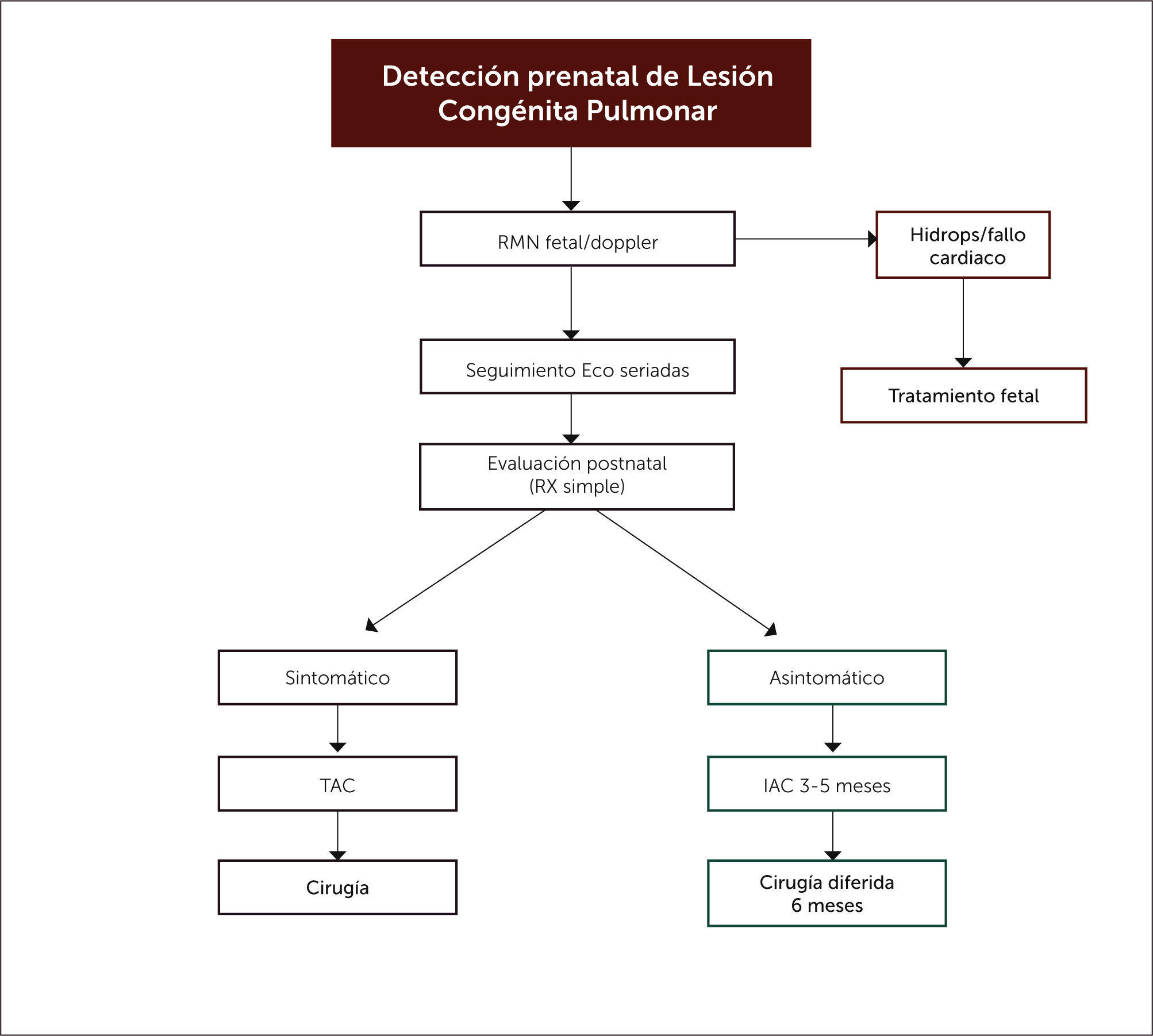
ENFRENTAMIENTO GENERAL DE LAS LESIONES PULMONARES CONGÉNITASFrente a la detección prenatal de una malformación pulmonar es relevante iniciar un seguimiento ecográfico estricto y complementar el estudio con doppler y RNM fetal que permitan orientar el diagnóstico diferencial e identificar aquellas en riesgo de desarrollar falla cardíaca e hidrops, las que pueden beneficiarse de una terapia prenatal. La lobectomía fetal es una terapia descrita para las lesiones sólidas más severas, pero tiene una tasa de sobrevida cercana al 50%, por lo que la cirugía durante el parto mediante un procedimiento EXIT se ha convertido en una alternativa con mejor sobrevida en la literatura1,38.
Otras opciones menos invasivas, ya mencionadas en el desarrollo de este capítulo, son la administración de corticoides maternos, aspiración percutánea de quistes, la derivación toracoamniótica, escleroterapia y la ablación con láser de la arteria nutricia. (Figura 9).
.")
Todas estas terapias requieren equipos médicos altamente especializados y deben realizarse sólo en centros experimentados38,58,60.
Respecto al enfrentamiento posnatal, no hay dudas en la indicación quirúrgica de los pacientes sintomáticos, independiente de la edad a la que se presenten los síntomas, pero existe alguna controversia sobre el momento oportuno para la cirugía en los pacientes asintomáticos, con grupos a favor de una conducta expectante, basados en estudios observacionales retrospectivos y en la involución espontánea intrauterina de algunas malformaciones pulmonares. Frente a esa postura, se encuentra la de aquellos grupos que han optado por una cirugía electiva ante el riesgo de infecciones recurrentes, neumotórax o malignización. La evidencia publicada muestra evolución postoperatoria favorable, bajas tasas de complicaciones y recuperación morfológica y funcional completa27,62. Nuestro centro basa su conducta en esta última postura, la que se refleja en el flujograma de evaluación y manejo de lesiones pulmonares congénitas mostrado en la figura 10.

El autor declara no tener conflictos de interés, en relación a este artículo.