




Se presenta en este artículo de revisión, un análisis de los factores a considerar en la toma de decisiones de manejo respecto a un grupo de pacientes portadores de cardiopatías congénitas complejas de muy alto riesgo, generalmente asociadas a comorbilidades y condiciones agravantes. Basándose en la experiencia de los autores, revisión de la literatura y casos clínicos ilustrativos, se sugieren posibilidades terapéuticas iniciales, poniendo énfasis en el análisis estrictamente caso a caso y las características que posea el centro de salud. Se proponen además nuevas posibilidades de tratamiento para el extremo del espectro en este grupo de recién nacidos, mostrando que una estrategia híbrida, quirúrgica-intervencional, es posible y segura para pacientes con bajo peso de nacimiento, anatomías extremadamente complejas o con comorbilidad asociada de alto riesgo. Para lograr éxito en este abordaje, es necesario que grupos multidisciplinarios se involucren en todo el proceso de diagnóstico y tratamiento.
This article will review, the decision making analysis, in a very selected group of high risk patients, being complex congenital heart disease, associated conditions and comorbidity the mean diagnostic features. Based on authors experience, literature review and clinical ilustrative cases, we suggest initial therapeutical options, highlighting the case by case and center related decision analysis. Novel strategies are proposed for the end of the spectrum in this group of newborn babies, showing that hybrid aproach is a feasible and safe alternative for patients with low birth weight, extremely complex anatomy, or high risk associated comorbidities. Multidisciplinary involvement is mandatory in the whole diagnosis and treatment process.
El recién nacido (RN) con cardiopatía congénita, es un paciente muy difícil de enfrentar, especialmente si se asocian comorbilidades de alto riesgo o prematurez1. Hoy en día los resultados de sobrevida en este grupo de pacientes son cada vez mejores y se están realizando grandes esfuerzos enfocados en aminorar las secuelas post quirúrgicas, siendo las más frecuentes y gravitantes las del sistema nervioso central (SNC)2. Cómo enfocaremos a estos pacientes tan complejos, que decisiones terapéuticas tomaremos y en qué momento se realizará alguna intervención serán los focos de nuestro análisis en este artículo. Además se revisarán aquellos factores que pueden causar impacto positivo en los resultados y se plantearán enfoques terapéuticos novedosos para enfrentar a estos pacientes.
MAGNITUD DEL PROBLEMA, CARACTERÍSTICAS DE LOS PACIENTES Y DIFICULTADES EN LA TOMA DE DECISIONESEl 0.8 a 1% de los recién nacidos nace con una cardiopatía congénita3. Las cardiopatías son la malformación congénita más frecuente y la segunda causa de muerte en los menores de un año en nuestro país. En Chile nacen aproximadamente 250000 niños cada año; de ellos, alrededor de 2000 serán portadores de una cardiopatía congénita al menos moderada. Estimamos que 180 niños requerirán cirugía en el periodo de RN y 35 pesarán menos de 2500 gramos y cada uno de ellos tiene sus propias características en relación a edad gestacional y peso. Existen 164 cardiopatías congénitas diferentes y 204 procedimientos distintos que se pueden realizar para tratar estas malformaciones4. Para complicar aún más las cosas, un 25% de ellos podrá tener una malformación congénita extra cardíaca asociada, siendo las más frecuentes las cromosomopatías5. Nos encontramos entonces frente a un problema con múltiples aristas; ¿cómo enfocamos a este paciente único? No existen algoritmos ni flujogramas que seguir en estas situaciones y por ello pensamos que para obtener los mejores resultados posibles, es necesario realizar un análisis de riesgo para cada paciente y situación, individualizando adecuadamente el problema y posteriormente enfocarlo con una mirada de equipo que permita un abordaje desde diferentes perspectivas. Se requiere de un equipo afiatado y multidisciplinario de neonatólogos, cardiólogos y cirujanos cardiovasculares así como también de otros subespecialistas de experiencia para realizar un análisis multifactorial y tomar las mejores decisiones. Frente a estos lábiles pacientes que además son únicos, nos debemos plantear las siguientes preguntas:
- 1.
¿Cuándo operamos?
- 2.
¿Qué alternativas quirúrgicas o intervencionales están disponibles?
- 3.
¿Realizar cirugía paliativa para mejorar los síntomas cardiovasculares y postergar la corrección propiamente tal?
- 4.
¿Un procedimiento híbrido, es decir colaborativo, quirúrgico y endovascular en conjunto con los cardiólogos-intervencionistas?
- 5.
¿O vamos directamente a la cirugía correctora?
Los pacientes RN, son más lábiles desde todo punto de vista. La madurez del sistema nervioso central (SNC), miocardio, pulmonar, renal, etc. no está completa, lo que los hace altamente susceptibles a cualquier noxa que ocurra en este período, además de poseer menos reservas que les permitan salir de situaciones fisiológicamente muy demandantes y extremas, como es el caso de la cirugía con circulación extracorpórea.
Avances en el manejo de la circulación extracorpórea (CEC), tales como miniaturización de los circuitos, menor grado de hemodilución y menor uso de hipotermia han hecho la cirugía neonatal con CEC, más segura reduciendo importantemente el sangrado postoperatorio y la respuesta inflamatoria sistémica. Hoy en día es seguro utilizar esta técnica en pacientes tan pequeños como 2kg, logrando buena protección miocárdica, tiempos de ventilación mecánica y hospitalización con postoperatorios cortos. Sin embargo, pese a estos adelantos la CEC sigue siendo una herramienta poco fisiológica con efectos clínicos notorios en estos pacientes6.
¿Entonces, por qué operamos a los RN de término con cardiopatías complejas en este período tan vulnerable?
¿Por qué no diferimos la cirugía?
Hay cardiopatías en las cuales, para que el RN esté estable, necesitamos mantener el ductus arterioso permeable, para lo cual utilizamos fármacos del tipo de las prostaglandinas (PG) sintéticas, las cuales tienen efectos secundarios significativos.
Estos pacientes además se encuentran en un equilibrio hemodinámico precario, con sobrecarga de volumen y/o presión, hipoxia y frecuentemente nos falta incluso un ventrículo, con sobrecirculación de sangre hacia los pulmones. Sabemos además que la cirugía cardiaca se puede realizar con buenos resultados y baja mortalidad, ya que esperar en estos pacientes con hemodinamia marginal los expone a patología intercurrente.
Otra indicación de cirugía es que aunque el paciente se encuentre en cierta estabilidad hemodinámica, la cardiopatía obliga a actuar en un plazo determinado de tiempo. Un ejemplo de esta situación es la transposición de las grandes arterias (TGA). Al nacer, los pacientes con TGA tienen ambos ventrículos igualmente desarrollados en cuanto a masa muscular. Esto se debe a que ambos ventrículos durante la vida intrauterina bombean contra una resistencia vascular similar. En la TGA el ventrículo izquierdo (VI) está conectado con el circuito pulmonar y al caer la resistencia vascular pulmonar empieza progresivamente a bombear contra una menor postcarga, lo que produce que este ventrículo disminuya su grosor y por lo tanto, su masa muscular. Al realizar el switch arterial, que es una reparación anatómica, se requiere que al reconectar el VI con la circulación sistémica, este tenga la fuerza suficiente para vencer la postcarga de esta circulación; si está adelgazado, no tendrá la potencia necesaria para vencer la resistencia. Por todo lo anterior, en TGA simple tenemos una ventana de 3 semanas, pero idealmente la operación debiera realizarse en los primeros 15 días.
IMPORTANCIA DEL DIAGNÓSTICO ANTENATAL Y EDAD GESTACIONALLa OMS acordó, hace aproximadamente 50 años, llamar RN de término a aquellos que nacían 3 semanas antes hasta 2 semanas después de las 40 semanas de gestación7.
Esta definición se basa en la presunción existente en aquella época de que no habría diferencias en morbimortalidad en los RN nacidos durante este periodo de 5 semanas. El RN debe enfrentar ajustes importantes en los primeros momentos de su vida extrauterina y sabemos ahora que en ello juega un papel muy importante su grado de maduración.
La maduración es un proceso continuo con cambios importantes incluso en las últimas semanas de gestación. Cambios sutiles pero importantes en el desarrollo del sistema respiratorio ocurren tardíamente en la gestación, incluyendo pasos críticos del oxido nítrico y producción de surfactante así como del clearance de líquido pulmonar fetal. Maduración incompleta de acumulación energética, función enzimática y de inmunidad, también contribuyen. Si en el RN sin cardiopatía hay diferencias significativas en los resultados de morbimortalidad, es lógico pensar que en aquellos portadores de cardiopatías congénitas esto sea mucho más gravitante aún.
El RN con cardiopatía tiene algunas peculiaridades que analizaremos a continuación y que explican lo importante que es en ellos la maduración completa. En estudios relativamente recientes se ha encontrado que pacientes con cardiopatías como TGA o ventrículos únicos tienen un desfase de maduración de su sistema nervioso central de aproximadamente un mes en comparación con RN sin cardiopatía congénita8,9. Esto se explicaría en parte por la fisiología de la circulación intrauterina. La circulación fetal se caracteriza por ser una circulación en paralelo y enviar un porcentaje mayoritario del débito cardiaco hacia la circulación sistémica en desmedro de la circulación pulmonar, la cual se reduce en forma importante por no haber necesidad de oxigenación y ausencia de ventilación. Si comparamos la saturación de oxígeno de la aorta fetal, esta es mayor que la saturación de la aorta torácica descendente. Se privilegia de esta forma que uno de los órganos más sensibles y que más se demora en desarrollar, el sistema nervioso, reciba sangre más oxigenada que el resto de los órganos10.
Unos de los grandes avances en la especialidad de la cardiología pediátrica ha sido el diagnóstico antenatal pues nos permite anticiparnos, determinar el lugar del parto en un centro adecuado y preparar a la familia. Además podemos articular un buen equipo multidisciplinario que reciba a este paciente, elemento esencial para una mejor evaluación y manejo del recién nacido. El cómo enfrentamos al paciente cardiópata desde el inicio es clave. Evitar que se cierre el ductus en cardiopatías ductus dependiente es un buen ejemplo. Si logramos anticiparnos y evitar a que un paciente caiga en shock cardiogénico o sufra de hipoxia severa lograremos disminuir la morbimortalidad preoperatoria y nos permite operar a los paciente en las mejores condiciones posibles y cuando lo necesiten.
Estudios recientes en pacientes con TGA han mostrado que con cada día que se demore la cirugía después de los 5 días iniciales de vida, se produce un aumento de costos y de morbilidad11.
Otros estudios han mostrado que la mortalidad preoperatoria de los pacientes con TGA es similar o mayor a la operatoria, y por lo tanto si queremos impactar en mejores resultados debemos optimizar el manejo desde el momento del nacimiento12.
Esto es particularmente importante en países con la geografía y organización de salud como la de Chile, en que los centros de manejo de pacientes de alta complejidad se encuentran centralizados en una sola ciudad y las distancias para acceder a ellos es muy grande.
El transporte de estos pacientes es muy complejo, requiere de aviones ambulancia y agrega riesgos importantes para el paciente y problemas sociales y familiares. Sin embargo, el diagnóstico antenatal también ha mostrado tener aspectos negativos.
La literatura ha sido categórica en mostrar que los RN con diagnóstico antenatal de cardiopatía congénita nacen 1 a 2 semanas antes en comparación con aquellos que no cuentan con diagnóstico antenatal. Las razones exactas son desconocidas, pero se citan como causas: cesárea electiva para mejor coordinación, ansiedad parental, preocupación del equipo médico (especialmente del equipo gineco-obstétrico) y falsos positivos de evaluaciones de bienestar fetal. En relación a esto se ha demostrado fehacientemente que la edad gestacional tiene un alto impacto en los resultados tanto de sobrevida como en estadía hospitalaria y costo, incluso en lo que se considera recién nacidos de término13,14.
Un estudio multicéntrico de 92 centros, analizó 4784 recién nacidos operados en un periodo de 2 años entre los años 2010 y 2011, obtenidos de la base de datos de la Sociedad Torácica de Cirujanos de Estados Unidos es muy revelador al respecto. Se realizó análisis multivariable de regresión logística para relacionar EG con mortalidad hospitalaria, tiempo de estadía y complicaciones ajustado a otras características de los pacientes. Como es de esperar, casi la mitad eran pacientes complejos como ventrículos únicos y TGA. Un 46% tenía diagnóstico antenatal, 31% eran RNT precoces y 48% nacieron antes de las 39 semanas. La mediana de la edad de cirugía fue de 7 días. Lo interesante e importante es que los mejores resultados de sobrevida se observaron en el grupo con una edad gestacional de 39.5 semanas con un 7.3% de mortalidad (rango: 6.3 a 8.4 para un límite de confianza de 95%). Para pacientes de 38 semanas subió a 8% y para aquellos de 37 semanas fue de 13.2%, lo que prácticamente duplica el riesgo de mortalidad. Resultados similares, todos estadísticamente significativos, se encontraron para largo de estadía postoperatoria y morbilidad. Por tal motivo debemos hacer todo lo posible para evitar el término electivo programado del parto a las 37 y 38 semanas en aquellos fetos con diagnóstico antenal de cardiopatía congénita, siempre y cuando las condiciones maternas y fetales lo permitan. La fecha de parto ideal es cerca de las 40 semanas. Este aspecto es necesario trabajarlo fuertemente con el equipo gineco-obstétrico, ya que con esta simple medida se logra mejorar significativamente los resultados de morbimortalidad. Suponemos que operar a recién nacidos más maduros también tendrá impacto en la futura evaluación neurológica de estos pacientes en el largo plazo. En estudios con resonancia nuclear magnética, se ha observado que recién nacidos operados de TGA hace 20 años, al ser evaluados en la adolescencia tienen una significativa mayor incidencia de lesiones focales y difusas cerebrales si las comparamos con la población normal y que aunque presentan un coeficiente intelectual similar al de la población general, tienen hasta cuatro veces mayor probabilidad de necesitar tutorías, educación diferencial o algún medicamento para tratamiento de salud mental15. Por lo tanto, la evidencia es clara al definir que la edad óptima para que nazca un feto con cardiopatía congénita es alrededor de las 39-40 semanas.
¿CÓMO ENFRENTAMOS AL RN QUE POR DISTINTAS RAZONES NACIÓ ANTES DE LAS 37 SEMANAS Y AL MENOR DE 2500 GRS?Estos pacientes por la inmadurez de sus órganos sumada a los trastornos hemodinámicos propios de su cardiopatía, los hace mucho más propensos a problemas neurológicos, infecciosos, respiratorios y a enterocolitis necrotizante por nombrar solo algunos de una larga lista de posibles problemas. Parece lógico asumir que al restaurar precozmente la fisiología cardiovascular, debiera mejorar la precaria estabilidad clínica de estos pacientes después de la cirugía. Por otro lado, se contrapone la tendencia natural a esperar mayor maduración y mejor peso para operar. El desafío en cada caso es encontrar el equilibrio entre operar y esperar mayor maduración En la toma de decisiones a este respecto van a influir la hemodinamia del paciente, la experiencia y filosofía de cada centro dentro de muchas otras variables16–18.
Es en estas situaciones donde se evidencia la relevancia de contar con un equipo multidisciplinario para tomar la mejor decisión.
¿Quiénes merecen la pregunta esperar vs operar?
Los pacientes que están inestables hemodinámicamente y requieren intervención pronta o aquellos en quienes no se puede operar por tener contraindicación como ECN o infección, no constituyen un gran dilema. La disyuntiva está en el paciente estable y poco invadido, especialmente si su peso es inferior a 2200 gramos. En ellos pensamos que demorar la reparación no confiere beneficio y se asocia a mayor morbilidad. Sobre todo si son sintomáticos cardiovascularmente, se recomienda la reparación temprana más que tratamiento médico y cirugía paliativa. Sin embargo, hay circunstancias en las cuales la cirugía correctora precoz no es la primera opción. Cuando hay estabilidad hemodinámica, en pacientes no conectados a ventilación mecánica, que suben de peso, no necesitan medicación cardiovascular o en pacientes con ventrículos únicos con hemodinamia estable con PG, es recomendable esperar un mejor peso y mayor maduración.
Resulta vital para las conductas más conservadoras contar con una unidad de recién nacidos que tenga tasas de infecciones muy bajas y prolijos cuidados de enfermería.
Por ejemplo, hemos manejado exitosamente a pacientes con ventrículo único prematuros extremos cerca de 3 meses con infusión de PG sin patología intercurrente. Estos resultados dependen fundamentalmente de las condiciones generales del centro y por lo tanto, no son extrapolables a otros servicios sino más bien representan resultados propios por lo que resulta muy difícil hacer recomendaciones específicas de cómo enfrentar a estos pacientes.
Algunos conceptos específicos:
- 1.
Hay patologías que no se benefician de cirugía paliativas como TGA, drenaje venoso pulmonar anómalo total.
- 2.
En menores de 1500 gramos portadores de cardiopatías con hiperflujo pulmonar, en especial comunicaciones interventriculares múltiples, la cirugía paliativa sí tendría un espacio.
- 3.
Intentar realizar cirugías que aumenten el flujo pulmonar (shunt de Blalock Taussig) es de muy alto riesgo en prematuros. Por tal motivo en el último tiempo y cada vez con más fuerza, se están utilizando paliaciones endovasculares en el laboratorio de cateterismo, como implantar un stent en el ductus para reemplazar el tratamiento prolongado con PGE. Más novedoso aún es implantarlo en el tracto de salida del ventrículo derecho con el propósito de mantener un flujo pulmonar adecuado mientras estos pacientes maduran y adquieren mayor peso, posponiendo así la cirugía. Estas constituyen nuevas modalidades de intervenciones paliativas que cada vez cobran mayor relevancia.
Paciente RNPT 29 semanas, diagnóstico antenatal de TGA con comunicación interventricular y atresia pulmonar, peso de nacimiento 1190 gramos, en buenas condiciones. Se inicia al nacimiento PGE1. Se logran saturaciones de 80-85% estables con buen estado hemodinámico.
Análisis: En este paciente, realizar una cirugía para aumentar el flujo pulmonar es de muy alto riesgo, así como lo es también el proceder a un implante endovascular de stent ductal, por el compromiso potencial de las ramas pulmonares con este tipo de terapias en este peso, y la fisiología de alto riesgo que genera la primera posibilidad en un paciente de este tamaño y madurez. Por este motivo se decide tomar conducta expectante, mantener PGE1 y esperar mayor peso y madurez para cirugía paliativa. Las principales preocupaciones serán disminuir al mínimo los riesgos de patologías intercurrentes, optimizar el manejo nutricional, evitar en lo posible necesidad de ventilación invasiva y manejo muy cuidadoso de accesos venosos evitando aumentar riesgo de trombosis u otros.
Este paciente se opera a los 3 meses de vida con peso de 2.9kg y se realiza una cirugía de Blalock-Taussig de 3.5mm, permanece hospitalizado 20 días post operatorios y se encuentra actualmente en espera de cirugía correctora a segunda etapa de paliación.
Paciente prematuro portador de transposición de grandes arteriasEste paciente también con diagnóstico antenatal de TGA, pesa 1700 gramos al nacer.
Análisis: En este caso, por la fisiología de su cardiopatía sólo se puede esperar algunas semanas antes de practicar cirugía correctora. Decidimos esperar ya que en su evaluación se determina que existe un foramen oval permeable adecuado y se inicia PGE1. Sin embargo, el aumento de peso no es el esperado ya que a los 16 días de vida sólo ha recuperado su peso de nacimiento luego de haber perdido un 10% inicialmente. Se decide realizar cirugía de switch arterial, la que con 1700 gramos es llevada a cabo bajo circulación extracorpórea, sin complicaciones, 72 horas de ventilación mecánica, requiere 16 días de hospitalización.
Recién nacido de pre término con obstrucción grave de la circulación sistémica y comunicación interventricularEste paciente nace de 34 semanas, sin diagnóstico antenatal de cardiopatía congénita en otro centro del país; pesa 1310 gramos y sus diagnósticos cardiológicos corresponden a una comunicación interventricular subaórtica con mal alineamiento posterior lo que genera una obstrucción subaórtica significativa. Además de una hipoplasia del arco aórtico con coartación aortica crítica, dependiente de PGE1.
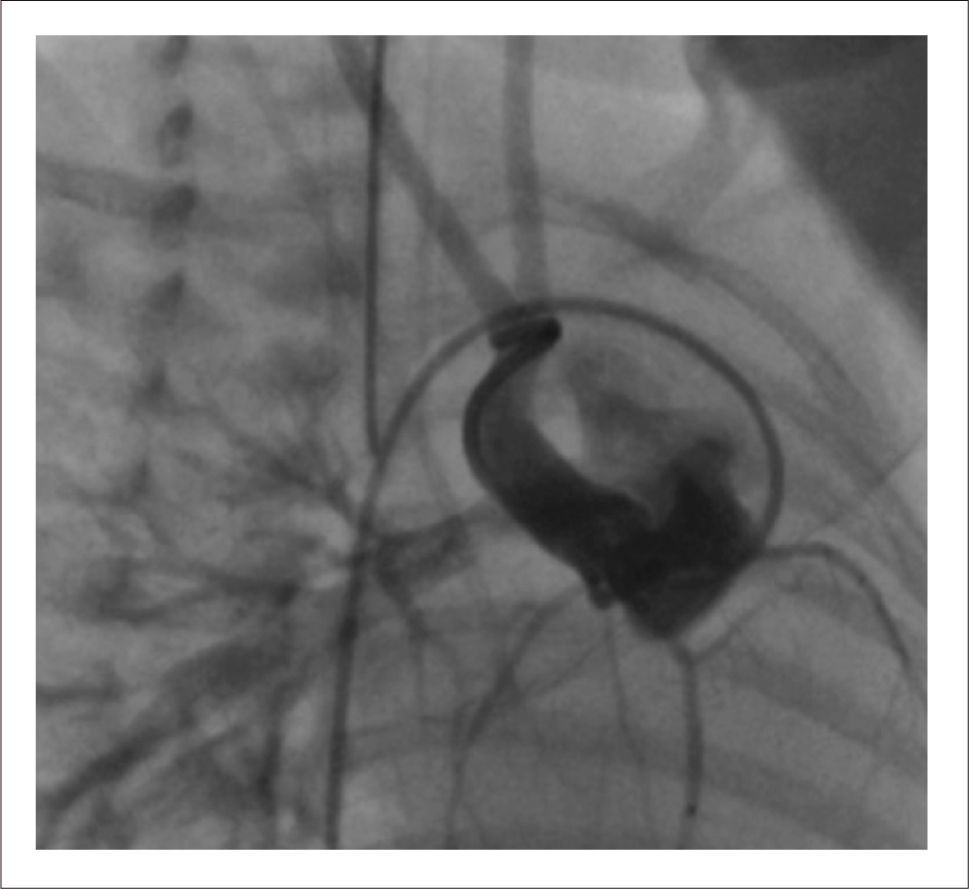
Análisis: Es derivada a nuestro centro para evaluación a los 2 días de vida y aquí el dilema es el de balancear el riesgo de una cirugía correctora de muy alto riesgo, cirugía paliativa también compleja y de muy alto riesgo en este peso (paliación tipo procedimiento de Norwood) vs mantener al paciente con infusión de PGE1 largo tiempo hospitalizado esperando peso adecuado para la corrección y por cierto, con fisiología inestable e insegura. La decisión del equipo fue la de realizar una paliación híbrida quirúrgica-intervencional. En pabellón quirúrgico se realiza bandings individuales a cada rama de la arteria pulmonar y luego en pabellón de hemodinamia se procede a instalación de stent en ductus arterioso de manera de mantenerlo permeable sin necesidad de uso de PG (Figura 1).

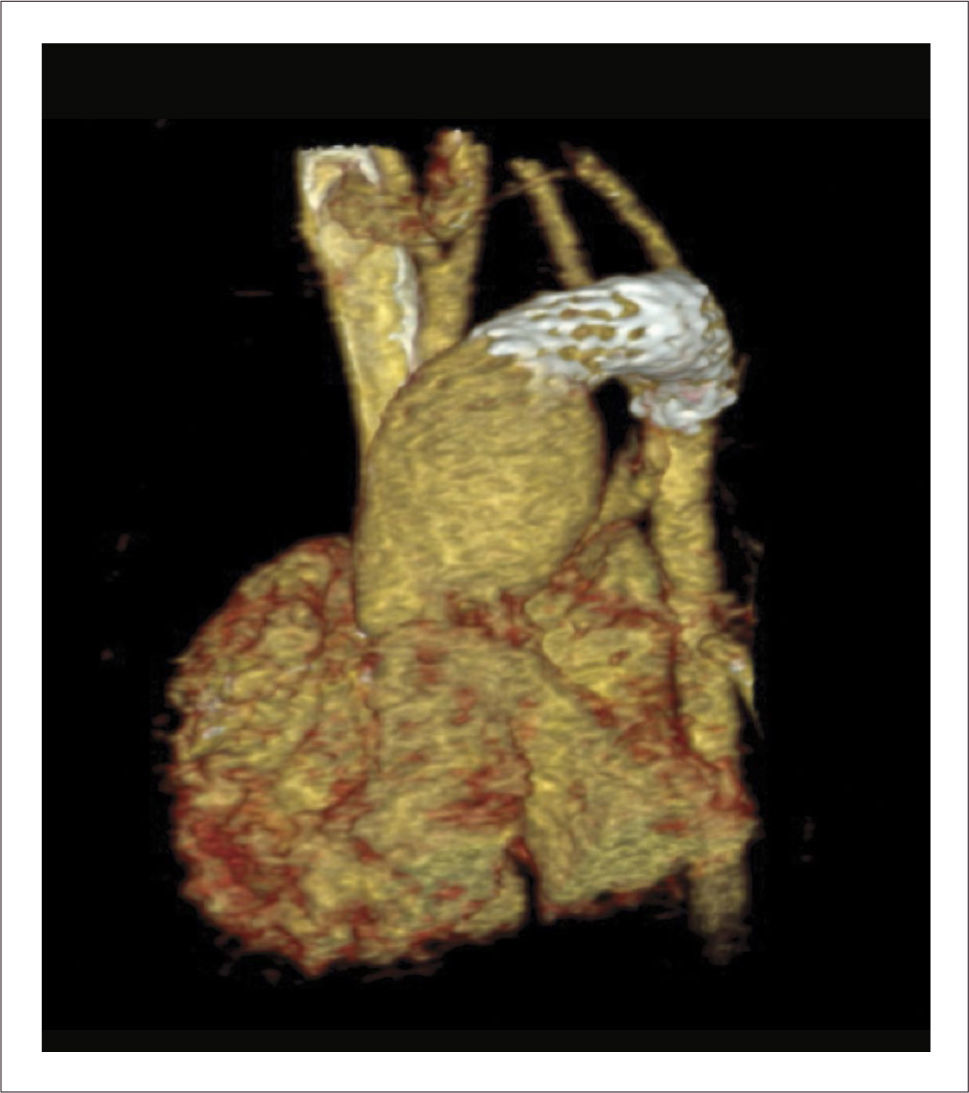
TOMOGRAFÍA AXIAL COMPUTADA ARCO AÓRTICO
Tomografía axial computada que muestra arco aórtico hipoplásico, con coartación crítica e hipoplasia del arco, post implante de stent ductal Banding de arteria pulmonar izquierda, y posterior a stent se ve parcialmente banding de arteria pulmonar derecha.
El paciente sale de pabellón con tórax cerrado y alta a los 5 días post implante de stent. Evolución posterior sin complicaciones, incrementos adecuados de peso y sin patología intercurrente. Evaluaciones ecocardiográficas posteriores y con TAC muestran crecimiento de aorta ascendente y mejoría del diámetro del anillo aórtico. Por lo que a los 4 meses de vida con 4.5kg de peso se somete a cirugía correctora, realizándose cierre de la CIV y reconstrucción del arco aórtico incluyendo parte del stent ductal en la reparación. En este caso posponer la cirugía con circulación extracorpórea permitió evitar el gran riesgo de daño sobre el SNC, que habría significado someter a este recién nacido pretérmino a paro circulatorio total hipotérmico o perfusión regional. Actualmente en muy buenas condiciones sin lesiones residuales significativas en ecocardiogramas de seguimiento.
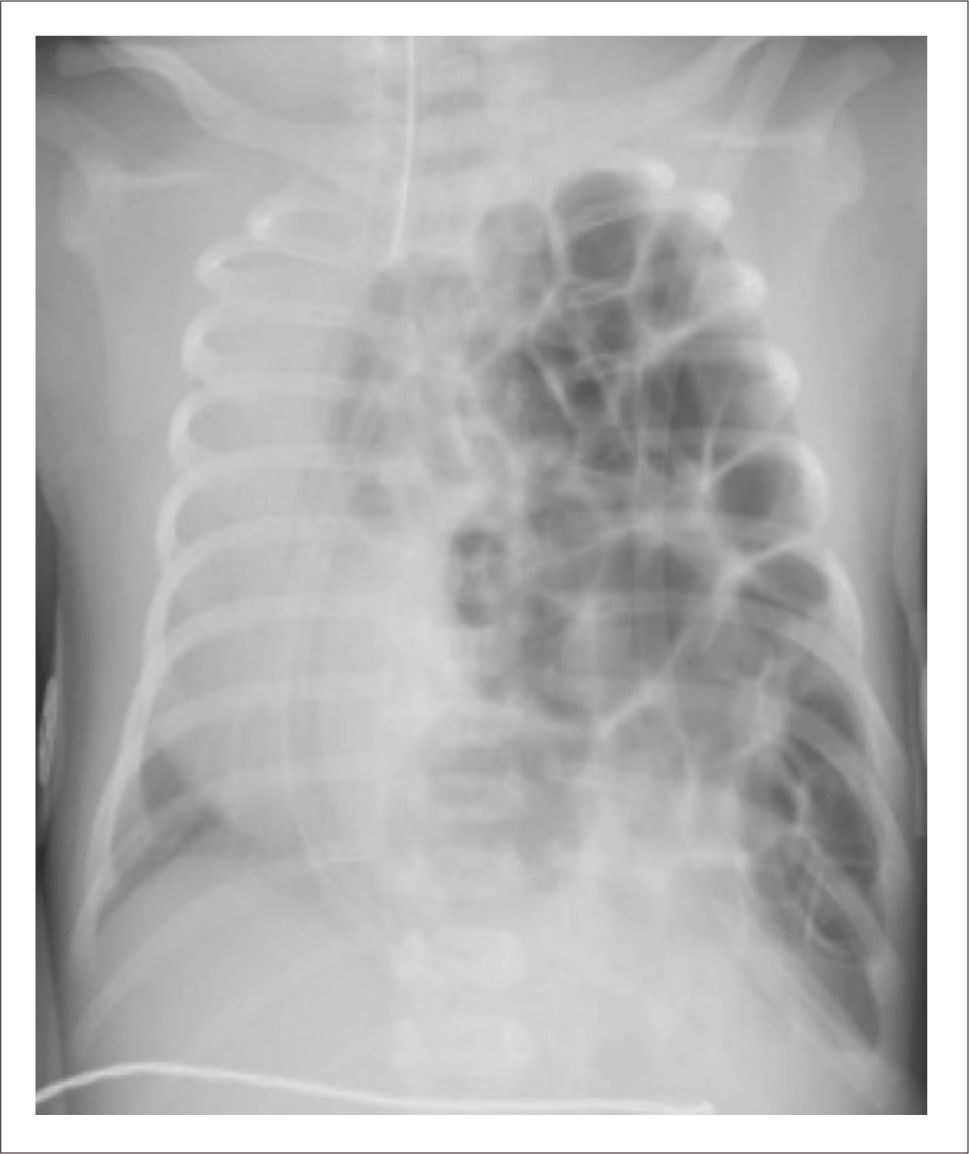
Recién nacido con diagnóstico de tronco arterioso, interrupción del arco aórtico y hernia diafragmática izquierda asociada a hipoplasia severa del pulmón ipsilateralRN de término, 3255 gramos. Anatomía: Tronco Arterioso. Válvula troncal cuadricúspide con insuficiencia moderada. Interrupción del arco aórtico tipo B. Hernia diafragmática izquierda severa, portador además de microdeleción del cromosoma 22q11.
Análisis pre-intervenciónSería muy dificultoso corregir esta patología en una etapa, ya que este caso involucra no solo una rara y compleja cardiopatía, sino también hipoplasia del pulmón izquierdo originada por el defecto diafragmático ipsilateral, sumado a la influencia que ejerce en un caso como este la elevada resistencia vascular pulmonar en el tipo de tratamiento a elegir, así como las comorbilidades del síndrome de Di George.
- I.
Primera etapa: Cirugía de la hernia diafragmática. Cuándo la RVP comienza a caer, pero aún elevada, se reparó la hernia (5to día de vida).
- II.
Procedimiento híbrido: Implante de stent ductal, en procedimiento percutáneo, de manera de evitar uso prolongado de PGE1 y realización posterior de bandings de arterias pulmonares, con niveles más bajos de RVP.
- III.
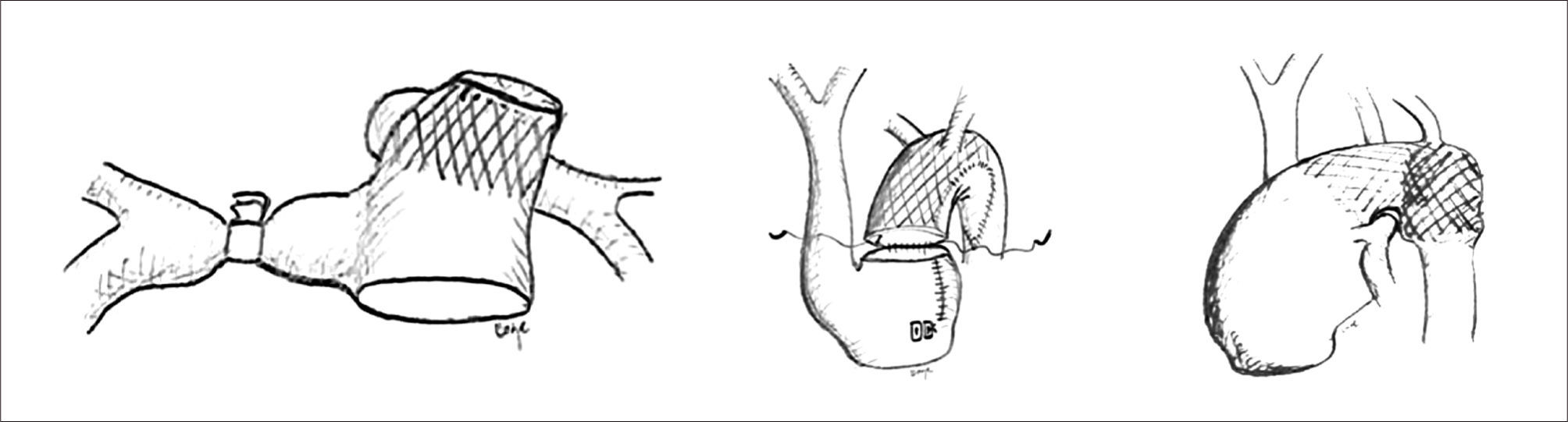
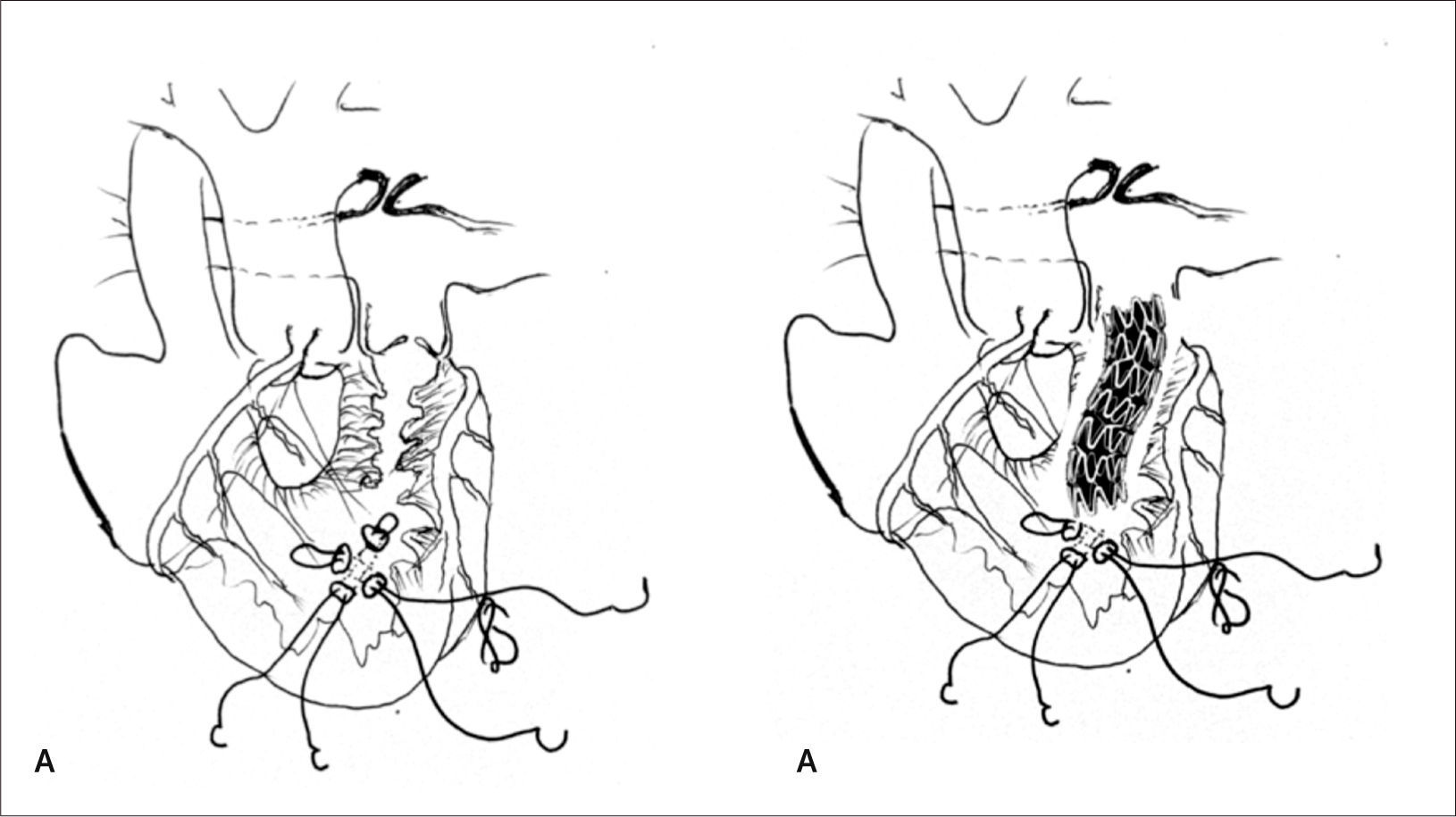
Corrección quirúrgica, postergada hasta los 5 meses de vida, permitiendo ganancia pondoestatural, y desarrollo pulmonar, especialmente izquierdo. Se cerró la CIV con parche de Dacron. Se reparó el arco aórtico incluyendo el stent ductal en parte de la plastía. De-banding de las arterias pulmonares y restablecimiento de la continuidad VD-APs con un conducto valvulado de bovino (Contegra® 12mm) (Figuras 2, 3, 4 y 5).




Este paciente nace a las 30 semanas de gestación, pesando 1300 gramos, con saturaciones mayores de 85% y con ecocardiografía que permite pensar en una buena posibilidad de éxito, al mantenerlo con infusión de PGE1, y esperar mayor peso para cirugía correctora en 1 etapa. Este periodo se programa cumplirlo en la unidad neonatal de origen. Sin embargo, evoluciona rápidamente con estenosis progresiva subpulmonar, que lo lleva a una situación crítica de hipoxia, por lo que debe ser trasladado a nuestro centro para tratamiento. El dilema en este caso es la realización de cirugía correctora en paciente de tan bajo peso vs realizar shunt aortopulmonar para mejorar hipoxia. Ambas alternativas nos parecen de muy alto riesgo, por este motivo se decide aumentar flujo pulmonar colocando un stent en el tracto de salida del ventrículo derecho de manera de eliminar el principal obstáculo en la circulación hacia el pulmón. Esto fue realizado en pabellón de hemodinamia, vía esternotomía, sin circulación extracorpórea, por punción directa perventricular del ventrículo derecho, se implanta stent en posición adecuada, saturaciones post procedimiento 92% (Figura 6). La evolución posterior fue marcada por hiperflujo pulmonar transitorio, manejado medicamente.

Alta hospitalaria a los 36 días. Se realiza cirugía correctora a los 5 meses de vida, con 5.2kg de peso. Se cierra la CIV y se amplia tracto de salida con parche transanular, retirando stent. Evolución post operatoria sin incidentes.
CONCLUSIONESEl proceso de toma de decisiones en este grupo de pacientes es extremadamente complejo, influido por múltiples factores, que hacen imposible aplicar un algoritmo. Debe analizarse caso a caso y establecer conductas para cada paciente específicamente, siempre considerando el contexto del centro donde nos desenvolvemos.
Cada paso que se da con estos pacientes, implica posiblemente tomar un determinado camino que no se podrá recorrer en sentido inverso, por lo tanto, avanzar en una dirección requiere necesariamente conocer en profundidad y tener la máxima experiencia posible en el paso siguiente.
Nos enfrentamos a un grupo de pacientes altamente seleccionado, algunos de ellos hasta hace muy poco se encontraban en una situación de muy baja probabilidad de éxito. Enfoques multidisciplinarios, colaboración cardiólogo-cirujano y apertura hacia la innovación, asociadas con un proceso prolijo y meticuloso de toma de decisiones, sin lugar a dudas mejorarán sus expectativas de sobrevida y especialmente de calidad de vida.
Los autores agradecen la enorme colaboración de los cirujanos cardiovasculares Drs. Guillermo Zamora, Francisco Boye y Luis Sánchez, en las cirugías de los pacientes presentados en este artículo. Ellos son además los autores de los diagramas que muestran técnicas quirúrgicas en detalle.
Los autores declaran no tener conflictos de interés, en relación a este artículo.