



Oxidative stress is a common feature in most hepatopathies. In recent years, evidence has accumulated that reactive oxygen species (ROS) induce a number of functional changes either deleterious or adaptive in the capability of the hepatocytes to produce bile and to secrete exogenous and endogenous compounds. This review is aimed to describe the mechanisms involved in these alterations. For this purpose, we will summarize:
- 1)
The current evidence that acutely-induced oxidative stress is cholestatic, by describing the mechanisms underlying the hepatocyte secretory failure, including the disorganization of the actin cytoskeleton and its most noticeable consequences, the impairment of tight-junctional structures and the endocytic internalization of canalicular transporters relevant to bile formation.
- 2)
The role for oxidative-stress-activated signalling pathways in the pathomechanisms described above, particularly those involving Ca2+ elevation and its consequent activation of Ca2+-dependent PKC isoforms.
- 3)
The mechanisms involved in the adaptive response against oxidative stress mediated by ROS-responsive transcription factors, involving up-regulation of GSH-synthesizing enzymes, GSH-detoxifying enzymes and the hepatocellular efflux pumps; this response enhances the co-coordinated inactivation by GSH conjugation of lipid peroxides and their further cellular extrusion.
- 4)
The manner this adaptive response can be surpassed by the sustained production of ROS, thus inducing transcriptional and posttranscriptional changes in transporters relevant to bile formation, as has been shown to occur, for example, after long-term administration of aluminum to rats, in the Long-Evans Cinnamon rat (a model of chronic hepatic copper accumulation mimicking Wilson’s disease), and in ischemia-reperfusion injury.
Reactive oxygen species (ROS) are formed during a variety of biochemical reactions and cellular functions. For example, leakage of electrons leading to superoxide anion (O2•—) generation may occur during the electron transport through the mitochondrial respiratory chain.1 Similarly, O2•— may be generated during the microsomal, cytochrome-P450-mediated hydroxylation of endo and exogenous compounds for detoxification. This is due to uncoupling of electrons during their transfer from NADPH cytochrome P450 reductase to cytochrome P450, or from cytochrome P450 to its substrates,2 as was shown to occur for ethanol3 and benzo(a)pyrene4 metabolisms, among many others. This steady-state ROS formation is balanced by a similar rate of consumption by the antioxidant defences provided by enzymes (e.g., superoxide dismutase, catalase, glutathione peroxidase, glutathione-5-transferase [GST]) and free-radical-scavenging endogenous and exogenous compounds (e.g., glutathione, ascorbate, α-tocopherol, β-carotene).5
Oxidative stress results from an imbalance between endogenous pro-oxidant formation and neutralization by antioxidant defences. When this occurs, ROS can damage virtually all types of biological molecules. These molecular alterations trigger a number of interrelated pathomechanisms, namely: i) free-thiol oxidation of proteins;6,7ii) disassembly of cytoskeleton,8,9 with plasma membrane blebbing as a major emergent;10iii) formation of mitochondrial permeability transition pores, leading to the release of mitocondrial promoters of apoptosis (cytochrome c, Smac/DIABLO) and uncoupling of the electron transport chain from ATP production, with eventual necrosis associated with ATP depletion;11iv) increased plasma-membrane peroxidation, leading to membrane permeabilization and leakage of cytosolic components;12v) oxidative breaking of DNA strands, leading to genotoxicity and carcinogenesis;13,14vi) elevation of intracellular Ca2+ levels, which facilitates mitochondrial permeability transition and activation of Ca2+-dependent proteases, endonucleases, and lipases.11,15
Due to its multiple energy-dependent functions, liver has a high mitochondrial metabolic rate, and is heavily engaged in detoxification mechanisms that involve redox-enzyme systems. Since these are major sources of endogenous free radicals, ROS production is higher in liver as compared with most organs. Hence, hepatocytes are equipped with abundant antioxidant enzymes to counterbalance this oxidative challenge.16 However, this borderline equilibrium makes liver highly susceptible to the pro-oxidant injury induced by pathological conditions that disrupt this balance, by enhancing ROS formation, by impairing the antioxidant defences, or both.17-19 Indeed, oxidative stress is a common feature in most hepatopathies including hepatic ischemia-reperfusion injury following hepatectomy or liver transplantation,20,21 obstructive cholestasis,22 chronic cholestatic liver diseases,23-27 sepsis-induced cholestasis,28 viral,29,30 toxic31 and autoimmune32 hepatitis, alcoholic30,33 and non-alcoholic34-36 steatohepatitis, and pathologies leading to hepatic accumulation of metals, such as iron (hemochromatosis, iron-loading anaemia)37,38 or copper (Wilson’s disease).39
Much attention has been focused on the mechanism of oxidative-stress-induced hepatocellular death.40-43 However, this usually involves only a limited number of hepatocytes. For example, in patients with hepatic oxidative stress due to chronic alcoholic intake, ~7 per 1000 hepatocytes suffer apoptosis,44 and only marginal necrosis has been observed in the liver of patients with a similar alcohol intake.45 The question therefore arises as to which extent the bulk of the remaining viable cells is functionally affected by the oxidative injury.
In recent years, evidence has accumulated that oxidative stress is cholestatic. Functional changes involve impairment of hepatocellular biliary secretory machinery through both the direct oxidative damage of relevant cellular structures by ROS or, indirectly, by modification of intracellular signal transduction pathways sensitive to changes in the intracellular redox state. This review is aimed to summarize the mechanisms involved in these alterations.
2. Mechanisms of hepatobiliary secretory function: «Let it bile»Bile formation is an osmotic process driven by the vectorial transport of certain solutes into bile. They comprise mainly bile salts and both oxidized (GSSG) and reduced (GSH) glutathione. For these solutes to induce blood-to-bile water transport primarily, they need to be concentrated and retained into a confined space (the bile canaliculus). The hermeticity is provided by tight-junctional structures localized in the paracellular pathway, near the apical surface of the cells.
Once secreted, these solutes induce passive water movement in response to osmotic gradients via both paracellular and transcellular routes. Transcellular water movements are facilitated by the water channels, aquaporins (AQP) type 9 and 8, located at the basolateral and apical membranes, respectively.46
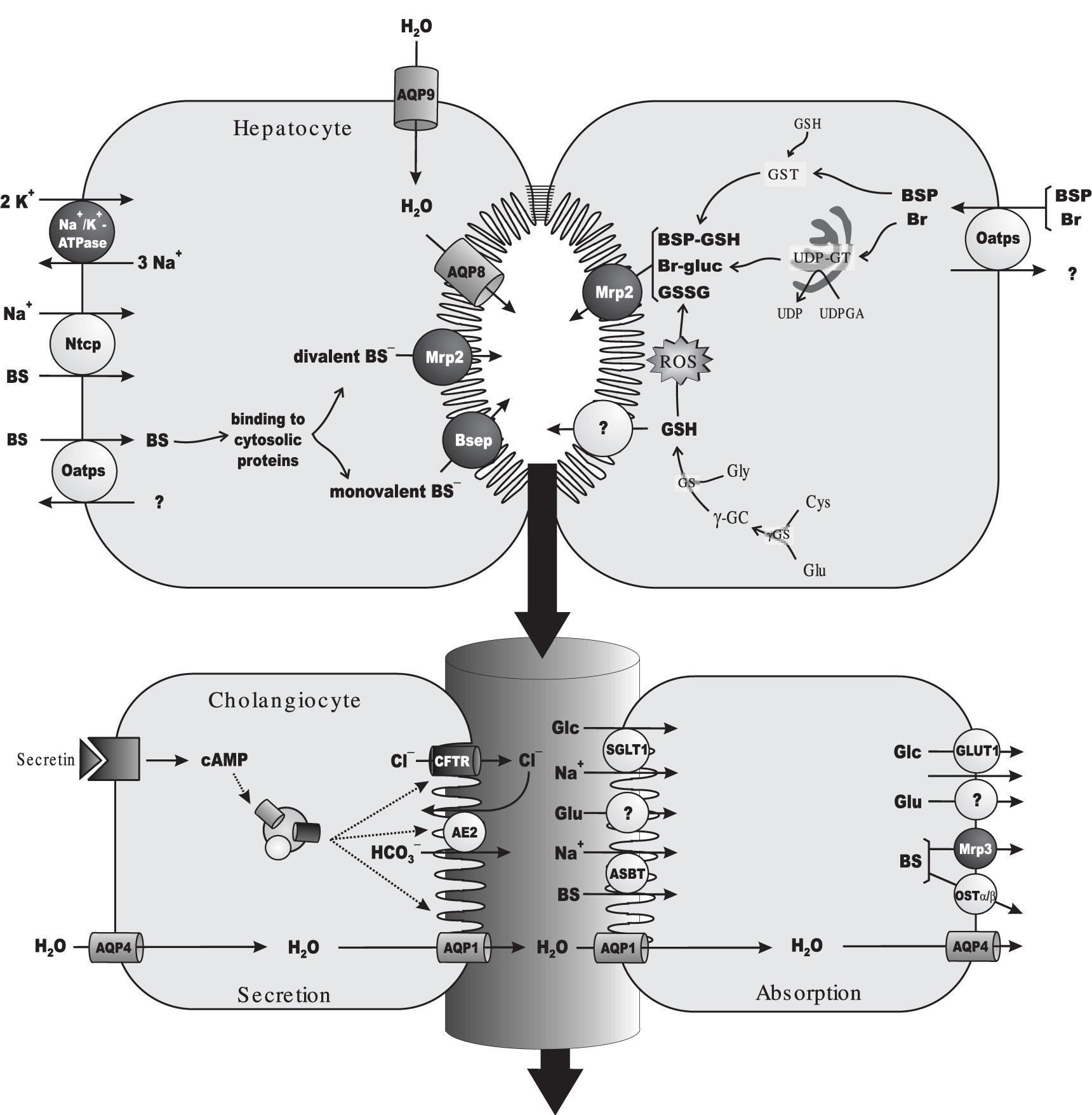
As expected for a solute exerting osmotic forces at the canalicular level, a linear relationship between bile salt secretion and bile flow has been observed in all vertebrates, including man. This bile flow fraction is conventionally referred to as «bile salt-dependent fraction of the bile flow».47 Since a positive value is, in most cases, obtained when extrapolating bile-salt output to zero, a bile salt-independent fraction of the bile flow is also apparent, which has been mainly attributed to glutathione output. This primary (canalicular) secretion is further modified by cholangiocytes during its transit along bile ducts, as a result of a balance between hormone-dependent water and electrolyte secretion and, on the other hand, the obligatory absorption of water, electrolytes and organic solutes.48,49Figure 1 depicts the main solutes and transporters relevant to bile formation at both the hepatocellular and the bile-ductular levels.
 and of the model exogenous cholephilic organic anion, sulphobromophthalein (BSP). Primary, ATP-dependent transporters are depicted in dark grey. For the sake of simplicity, only rodent transporters are shown, but human orthologs have been identified for all of them (see section 2 for details). Na+-dependent uptake of bile salts (BSs) at the sinusoidal level is mediated by the Na+-taurocholate cotransporting polypeptide (Ntcp), driven by the Na+ gradient generated by Na+/K+-ATPase. BSs are also taken up in a Na+-independent manner by the organic anion-transporting polypeptide (Oatp) family of transporters, which transport a wide range of solutes, including endogenous and exogenous organic anions, such as unconjugated Br and BSP. After traversing the cell bound to cytosolic protein, taurine or glycine-conjugated, monovalent BSs are excreted at the canalicular pole by the bile salt export pump (Bsep). Sulfated or glucuronidated, divalent BSs are instead transported by the multidrug resistance-associated protein 2 (Mrp2). Following their Oatp-mediated uptake, Br is conjugated with uridine diphosphate (UDP)-glucuronic acid (UDPGA) by the endoplasmic-reticulum enzyme, UDP-glucuronosyltransferase (UDPGT), to form Br mono/di-glucuronides (Br-gluc), which are excreted by Mrp2. BSP is conjugated with reduced glutathione (GSH) by the cytosolic enzyme, glutathione 5-transferase, to form the glutathione-conjugated parent compound BSP-GSH, which can be also excreted by Mrp2. GSH available for biliary excretion is provided by de novo synthesis. GSH biosynthesis comprises a two-step reaction catalyzed by the enzymes, γ-glutamyl-cysteinyl synthetase (γ-GCS) and GSH synthetase (GS). γ-GCS catalyzes the formation of L-γ-glutamyl-cysteine (γ- GC) from L-glutamate (Glu) and L-cysteine (Cys), whereas GS catalyzes GSH synthesis by further incorporation of L-glycine (Gly). GSH is excreted with high affinity by an as yet unidentified high-affinity canalicular GSH transporter, whereas its oxidized form, GSSG, is excreted with high affinity by Mrp2. Some cross-transport may however exist, particularly when any of these transporters is saturated (not shown). Both BSs and GSH/GSSG can act as primary driven force for osmotic bile formation, by promoting water movements via AQP9 and AQP8, localized at the sinusoidal and canalicular membrane domains, respectively. Canalicular bile flow is further modified during its transit along the bile ducts by both secretory and absorptive processes. Ductular fluid secretion is driven by the secretin-regulated, cAMP-dependent output of a HCO3--rich fluid facilitated by the anion exchanger 2 (Ae2). Exchange is driven by the out-to-in Cl--concentration gradient created by the ATP-dependent, secretin-activated channel, cystic fibrosis transmembrane regulator (CFTR). Blood-to-bile water movement is facilitated by constitutive AQP4 in the basolateral membrane, and secretin-stimulated AQP1 in the apical membrane. Absorption is driven by the osmotic gradients created by Glu (through unidentified transporters), glucose (Glc; transported by SGLT1 and GLUT1 at the apical and basolateral domains, respectively) and BSs (taken up by the apical Na+-dependent bile salt transporter, Asbt, followed by basolateral extrusion via the multidrug resistance-associated protein 3, Mrp3, and the heterodimeric organic solute transporter, Ostα-Osβ).")
Main transport proteins and metabolic events involved in bile-flow generation and in the hepatic handling of the endogenous organic anion, bilirubin (Br) and of the model exogenous cholephilic organic anion, sulphobromophthalein (BSP). Primary, ATP-dependent transporters are depicted in dark grey. For the sake of simplicity, only rodent transporters are shown, but human orthologs have been identified for all of them (see section 2 for details). Na+-dependent uptake of bile salts (BSs) at the sinusoidal level is mediated by the Na+-taurocholate cotransporting polypeptide (Ntcp), driven by the Na+ gradient generated by Na+/K+-ATPase. BSs are also taken up in a Na+-independent manner by the organic anion-transporting polypeptide (Oatp) family of transporters, which transport a wide range of solutes, including endogenous and exogenous organic anions, such as unconjugated Br and BSP. After traversing the cell bound to cytosolic protein, taurine or glycine-conjugated, monovalent BSs are excreted at the canalicular pole by the bile salt export pump (Bsep). Sulfated or glucuronidated, divalent BSs are instead transported by the multidrug resistance-associated protein 2 (Mrp2). Following their Oatp-mediated uptake, Br is conjugated with uridine diphosphate (UDP)-glucuronic acid (UDPGA) by the endoplasmic-reticulum enzyme, UDP-glucuronosyltransferase (UDPGT), to form Br mono/di-glucuronides (Br-gluc), which are excreted by Mrp2. BSP is conjugated with reduced glutathione (GSH) by the cytosolic enzyme, glutathione 5-transferase, to form the glutathione-conjugated parent compound BSP-GSH, which can be also excreted by Mrp2. GSH available for biliary excretion is provided by de novo synthesis. GSH biosynthesis comprises a two-step reaction catalyzed by the enzymes, γ-glutamyl-cysteinyl synthetase (γ-GCS) and GSH synthetase (GS). γ-GCS catalyzes the formation of L-γ-glutamyl-cysteine (γ- GC) from L-glutamate (Glu) and L-cysteine (Cys), whereas GS catalyzes GSH synthesis by further incorporation of L-glycine (Gly). GSH is excreted with high affinity by an as yet unidentified high-affinity canalicular GSH transporter, whereas its oxidized form, GSSG, is excreted with high affinity by Mrp2. Some cross-transport may however exist, particularly when any of these transporters is saturated (not shown). Both BSs and GSH/GSSG can act as primary driven force for osmotic bile formation, by promoting water movements via AQP9 and AQP8, localized at the sinusoidal and canalicular membrane domains, respectively. Canalicular bile flow is further modified during its transit along the bile ducts by both secretory and absorptive processes. Ductular fluid secretion is driven by the secretin-regulated, cAMP-dependent output of a HCO3--rich fluid facilitated by the anion exchanger 2 (Ae2). Exchange is driven by the out-to-in Cl--concentration gradient created by the ATP-dependent, secretin-activated channel, cystic fibrosis transmembrane regulator (CFTR). Blood-to-bile water movement is facilitated by constitutive AQP4 in the basolateral membrane, and secretin-stimulated AQP1 in the apical membrane. Absorption is driven by the osmotic gradients created by Glu (through unidentified transporters), glucose (Glc; transported by SGLT1 and GLUT1 at the apical and basolateral domains, respectively) and BSs (taken up by the apical Na+-dependent bile salt transporter, Asbt, followed by basolateral extrusion via the multidrug resistance-associated protein 3, Mrp3, and the heterodimeric organic solute transporter, Ostα-Osβ).
Bile salts are the predominant organic solutes in bile. The main sinusoidal transport system for bile-salt uptake is the Na+-taurocholate cotransporting polypeptide, which has been cloned from both rat (Ntcp, Slc10a1)50 and human liver (NTCP, SLC10A1).51 Ntcp/NTCP is driven by a transmembrane Na+ gradient maintained by the Na+/K+-ATPase pump, which is also strategically localized in the sinusoidal membrane.52 Ntcp/NTCP accounts for the transport of more than 80% of amidated bile salts (the major circulating bile salts), and only 40% of their unconjugated, parent compounds.53 The remaining fraction of circulating bile salts is taken up by a non-electrogenic, Na+-independent transport system, formed by a family of transporters collectively named organic aniontransporting polypeptides (Oatps/OATPs for rat and human, respectively).54 Humans possess four OATPs: OATP1A2 (SLCO1A2/SLC21A3), OATP1B1 (SLC21A6), OATP1B3 (SLC21A8) and OATP2B1 (SLC21A9). In the rat, there are three ones: Oatp1a1 (Slc21a1), Oatp1a4 (Slc21a5) and Oatp1b2 (Slc21a10). Apart from bile salts, Oatps/OATPs accept a wide range of amphipathic, organic compounds, including bilirubin, bilirubin glucuronides, leukotrienes, estrogens, «type II» organic cations and several exogenous organic anions, with the cholephilic dye sulphobromophthalein (BSP) being a prototypical example of the latter ones.55 Certain specificity exists among Oatp isoforms. All but OATP2B1 are involved in bile salt uptake in humans,56 whereas bilirubin diglucuronides and, perhaps, unconjugated bilirubin are transported only by OATP1B1.57 In rats, all of the iso forms are involved in uptake of conjugated and unconjugated bile salts,56 whereas only Oatplal and Oatp1a4 are involved in bilirubin monoglucuronide transport.58
After traversing the cell by Fick’s diffusion bound to high-affinity cytosolic proteins, monoanionic bile salts (C24 amides conjugated with glycine or taurine) are excreted in the canalicular pole by the bile salt export pump (BSEP/Bsep), an ATP-binding cassette transporter.59 In contrast, canalicular efflux of divalent, bipolar sulphated or glucuronidated bile salts is mediated by the multidrug resistance-associated protein 2 (MRP2/Mrp2; ABCC2/Abcc2). This carrier is also engaged in the biliary excretion of endogenous and exogenous non-bileacid organic anions conjugated with glutathione, glucuronic acid or sulphate. They comprise leukotriene C4, bilirubin glucuronides, some steroid sulphates and cholephilic dyes, including BSP both in its unconjugated and glutathione-conjugated forms.60
The bile-salt-independent fraction of the bile flow depends on glutathione excretion, mainly in its reduced form, GSH (~80%).61,62 Hepatocellular glutathione transport mechanisms are poorly understood. The liver is the main site of glutathione synthesis, exporting this peptide into both blood and bile. Most, if not all biliary glutathione comes from this intracellular source.63 No canalicular transporter for glutathione has been cloned so far. However, a high-affinity, electrogenic carrier has been functionally characterized.64-67 This transporter exports actively GSH into bile, and can transfer GSSG and GSH conjugates as well, but with lower affinity. Another transporter likely involved in glutathione canalicular transport is Mrp2. However, this carrier bears low affinity towards GSH, although it can transfers GSSG and GSH-conjugates with high affinity.65
Canalicular bile flow is further modified during its transit along bile ducts by both secretory and absorptive processes.68 Ductular fluid secretion is mainly driven by the secretin-regulated, cAMP-dependent output of a HCO3--rich fluid facilitated by the Cl-/HCO3--exchange system, anion exchanger 2 (Ae2/AE2). Exchange is dependent on the out-to-in Cl-concentration gradient, which is maintained by the Cl- efflux across the apical membrane via the ATP-dependent, secretin-activated, cystic fibrosis transmembrane regulator (CFTR). Blood-to-bile water movement at the ductular level is facilitated by constitutive AQP4 in the basolateral membrane, and secretin-stimulated AQP1 in the apical membrane.46 On the other hand, absorption of ductular water and electrolytes is driven by the osmotic gradients created by bile-to-plasma transport of electrolytes and organic solutes. They comprise: i) glutamate, transported by as yet unidentified carriers; ii) glucose, transported by SGLT1 and GLUT1 at the apical and basolateral domains, respectively; iii) bile salts, taken up by the apical Na+-dependent bile salt transporter, ASBT/Asbt (SLC10A2/slc10a2), and extruded by both the basolateral export pump, MRP3/Mrp3 (ABCC3/Abcc3), and the heterodimeric organic solute transporter, OSTα-OSTβ/Ostα-Ostβ.49,69
3. Changes in hepatobiliary secretory function induced by oxidative stressCompelling evidence in the literature indicates that oxidative challenge affects the hepatocyte-secretory machinery by impairing both bile flow (hepatocellular cholestasis) and the biliary excretion of endo and xenobiotics.
Oxidative-stress-induced impairment of bile flow generation has been demonstrated to occur after exposure to a number of pro-oxidant agents, including. tert-butylhydroperoxide (tBOOH),70,71 hydrogen peroxide,70,72 menadione,73 allyl alcohol,73 ethylhexanol,73 chloro-dinitrobenzene,71 CCl4,74 ethacrynic acid75 and lindane,76 among others. Some pharmacological agents, such as cyclosporine A,77 dapsone78,79 and nitrofuran derivatives,80,81 also induce cholestasis due, at least in part, to their prooxidant properties. Finally, maneuvers leading to hepatic oxidative stress, such as hepatic82,83-87 and intestinal88 ischemia-reperfusion, or aluminum intoxication,89,90 also induce bile flow impairment.
These cholestatic conditions are often associated with impairment of the bile salt-dependent bile flow. In many of these cases, the decrease in the overall bile flow was apparent even when the bile salt-independent bile flow was enhanced due to augmented GSSG output; GSSG extrusion into bile helps to maintain the intracellular GSH/GSSG ratio constant, and hence the intracellular redox equilibrium.70,91 However, under milder oxidative stress conditions, total bile flow may not change. In this case, the decrease in the bile salt-dependent bile flow induced by ROS may not be enough to counteract the parallel increase in the bile salt-independent bile flow due to enhanced GSSG biliary excretion.70,92 Similarly, osmotic choleresis associated with the biliary excretion of metabolites of the pro-oxidant agents used may mask ROS-induced cholestasis, as occurs soon after the administration of ethacrynic acid.70,75,93 However, we can consider these situations as cholestatic as well, since at least part of the hepatocyte machinery involved in bile formation is impaired.
3.1. Impairment of the hepatobiliary function by oxidative stress: «The orthodox view»The classical view to interpret the cholestasis associated to pro-oxidant conditions is based upon the occurrence of the following pathomechanisms:
- i)
Oxidative-stress-induced hepatocellular cell death by necrosis41 and apoptosis,42,94 which produces a reduction of the number of living, functional parenchymal cells. This pathomechanism is mainly associated with Ca2+-facilitated mitochondrial permeability transition, followed by release of mitochondrial pro-apoptotic factors and impairment of the electron transport chain, leading eventually to ATP depletion and necrotic cell death.95•—96
- ii)
Impairment of the bile salt-dependent fraction of the bile flow due to competitive inhibition of bile salt transport by the intracellular GSSG formed in excess during the oxidative challenge.70,72 This is supported by the fact that GSSG cis-inhibits the transport of the model bile salt, taurocholate, in liver canalicular membrane vesicles.97 In line with this contention, mirror curves depicting an increase in biliary excretion of GSSG and a decrease in biliary excretion of bile salts were obtained by administrating different pro-oxidant compounds in the isolated rat perfused liver, such as hydrogen peroxide,72 menadione92 and tBOOH.70,72 Furthermore, an inverse relationship between intracellular GSSG content and the percentage of inhibition of the biliary bile salt output was obtained by manipulating GSSG content using different oxidizing compounds, in the presence or absence of inhibitors of GSSG formation.72,92
- iii)
Impairment of the bile salt-independent bile flow due to a decrease in the biliary excretion of total glutathione (GSH plus GSSG). This occurs as a consequence of a depletion of the hepatic levels of glutathione due to the sustained plasmatic and biliary exportation from the cell as GSSG to maintain the GSH/ GSSG ratio.83
The above-mentioned mechanisms may be predominant under strong oxidizing conditions. However, it has become increasingly apparent that under mild, or even low, oxidizing conditions, functional alterations in the hepatocyte capability to produce bile may occur. Indeed, oxidative stress-induced drop of bile flow and/or decrease of the bile salt-secretory rate becomes aparent before leakage of cytosolic hepatocellular enzymes or increments in intracellular GSSG levels is induced by administration of different pro-oxidant compounds to isolated perfused rat livers.70,75 Likewise, in isolated rat hepatocyte couplets, an in vitro model for the study of hepatocanalicular function, the secretion of fluorescent bile-salt analogues into the canalicular vacuole was impaired by low concentrations of the pro-oxidant compounds, tBOOH and 2,3-dimethoxy-1,4-naphthoquinone, even when GSSG intracellular levels and cellular viability were unaffected.98 Finally, also in the couplet model, a dissociation between apical secretion of fluorescent bile-salt analogues and the amount of GSSG produced in the cell by the oxidizing effects of redox-cycling quinones has been reported.99 Overall, these results suggest that more subtle changes in the machinery involved in bile formation occur under mild oxidative stress conditions. The actin cytoskeletal disruption, which occurs even at very low oxidative stress levels, seems to be a crucial, causal factor.
The actin cytoskeleton is a dynamic network of filamentous actin (F-actin), formed by reversible assembly of monomeric actin (G-actin). It plays a crucial role in cell motility and in cell-shape changes occurring during the cellular cycle.100 In addition, actin is essential for the control of cell-cell interactions.100 For example, F-actin is anchored to Zonula occludens-associated proteins, such as ZO-1, ZO-2, ZO-3, 7H6 and cingulin, thus regulating the paracellular permeability by changes in its contractility status/organization.101 Actin is also involved in transcitosis processes, by operating as a ‘bridge’ between microtubules and the apical membrane itself, in a coordinated action of the microtubule-and the F-actin-based motor proteins, kinesin and myosin, respectively.102 Actin can also interact with, and possibly regulate, transmembrane proteins via binding to interacting-partner proteins, such as PDZ and HAX-1. These cytoskeleton-associated proteins are required for the biosynthetic targeting of transmembrane proteins from the trans-Golgi network to the proper membrane domain, and for their further cell-surface retention.103-105
The actin cytoskeleton is one of the primary targets of oxidative stress.8 The oxidative challenge promotes the oxidation of actin at a sulfhydryl group of cysteine in position 374.8 This induces conspicuous changes in actinspatial distribution, resulting in marked changes in the cellular topology (plasma membrane blebbing).8,106
Most of the above-mentioned roles of actin in cellular biology apply to hepatocytes, and many of them are involved in biliary secretory processes. It is therefore not surprising that disorganization of the actin cytoskeleton induced by oxidative stress has several deleterious effects on hepatobiliary function.
The interrelationship between oxidative stress, Ca2+ elevations, actin-cytoskeletal integrity and hepatocanalicular secretory function was exhaustively investigated in the 90‘s by Prof. Coleman’s laboratory, using the hepatocyte couplet model. These studies revealed that, under mild oxidative stress conditions with preserved hepatocellular viability, a close relationship exists between the impairment in the capability of couplets to accumulate apically and retain in their canalicular vacuoles fluorescent bile-salt analogues and the disarrangement of the pericanalicular actin cytoskeleton induced by the oxidizing compounds, tBOOH98,107-109 and menadione.99,110,111 These two independent tests indicated that both the apical secretion of bile salts and their further tight-junctional-dependent retention in the bile canaliculus are impaired early under oxidative stress conditions. Essentially, the same findings were obtained in isolated perfused rat livers. When acutely administered in the perfusate at non-necrotic concentrations, tBOOH impaired bile salt excretion and increased the bile/perfusate ratio of [14C]- sucrose, a tight-junctional-integrity marker that enters the bile canaliculus via the paracellular pathway.70 The impairment in bile salt excretion and the disruption of the tight-junctional barrier function may be causally associated, since the latter induces leakage of bile salts (or other osmotically-active biliary solutes) from the canalicular space via the paracellular pathway, leading to dissipation of the osmotic gradients involved in bile formation.112
These functional alterations to secrete and retain bile salts in the biliary space seem to have a structural correlate. tBOOH induces disorganization of the tight-junctional complex in hepatocyte couplets, as suggested by the redistribution of the tight-junctional-associated protein, ZO-1.98 Changes in ZO-1 localization are however not observed in isolated perfused rat livers exposed to tBOOH, suggesting that the couplet model is a more sensitive model to study associations between functional and structural changes of the tight-junctional barrier. In addition, the canalicular bile salt transporter, Bsep, suffers endocytic internalization into intracellular vesicles in hepatocyte couplets,109 which reduces dramatically the density of transporters properly located at the membrane domain. A similar phenomenon has been described for Mrp2, another canalicular transporter relevant to bile formation. Indeed, retrieval of Mrp2 from the canalicular membrane into subapical vesicles occurs after exposure of isolated perfused rat livers to the pro-oxidant agents, tBOOH,71 chloro-dinitrobenzene71 and ethacrynic acid,75,113 or after hepatic ischemia-reperfusion.114 Mrp2 relocalization has a clear-cut functional correlate. Experiments in isolated perfused rat livers indicated that a high, sustained exposure to ethacrynic acid has an inhibitory effect on the excretion of both unchanged and conjugated forms of the model cholephilic dye, BSP.115 Haemodynamic changes induced by oxidative stress may also affect BSP hepatic handling, but the source of ROS should be extracellular rather than intracellular to induce this effect. Perfusion with a medium containing hypoxanthine in the presence of xanthine oxidase, which catalyzes in situ the conversion of hypoxanthine to O2•— and uric acid, increased hepatic vascular resistance in isolated rat livers, which is accompanied with a decrease in BSP uptake.116
The exact mechanisms that link oxidative-stress-induced actin disorganization with tight-junctional impairment and transporter internalization are unknown, but previous studies in the literature provide some clues. Hepatic tight-junctional permeability increases following actin-cytoskeleton disruption induced by phalloidin, a toxin that induces actin disorganization by binding specifically to F-actin and impeding its depolymerization.117 As indicated above, F-actin is anchored to tightjunctional-associated proteins (e.g., ZO-1, cingulin), and it is likely that F-actin-conformational disorganization induces relocalization of these intermediary proteins as well. In addition, F-actin disorganization may alter localization and/or occlusive function of proteins forming the tight-junctional strands, such as occludin and claudins. For example, ZO-1118 and cingulin119 bind to occludin apart from F-actin, and both tight-junctional-associated proteins interact with claudins as well.120 At present, the functional relevance of all these interactions remains speculative, and a clearer picture of the mechanisms involved in the actin-mediated oxidative tight-junctional impairment awaits further characterization.
In addition to impairing tight-junctional permeability, phalloidin-induced F-actin disorganization also induces internalization of canalicular transporters. This was shown to occur for Mrp2 and for one or more canalicular transporters belonging to the P-glycoprotein family (i.e., Mdr1a, Mdr1b, Mdr2 and Bsep).121 The retrieval of canalicular transporters under oxidative-stress conditions71,75,109,113 is therefore also likely due to the simultaneous F-actin disarrangement. The molecular bases to understand this causal relationship are just emerging. Mice lacking radixin, which links actin filaments to plasma-membrane proteins, develop conjugated hyperbilirubinemia associated to retrieval of Mrp2,122 and the same hols true for obstructive and estrogen-induced cholestasis, where a disturbed colocalization of Mrp2 and radixin is associated with Mrp2 endocytic internalization.123 Interestingly, the internalization of Mrp2 that occurs after hepatic ischemia-reperfusion is coincident with a virtual loss of radixin expression.124 Mrp2 retention in the apical membrane also requires interaction with the PDZ-domain protein, PDZK1.125 Since other canalicular transporters, such as Bsep, Mdr1 and Mdr2, do not contain obvious PDZ-interacting motifs, regulation by actin of the localization of these transporters must occur through a different subset of proteins. A likely candidate is HAX-1, a cytoskeleton-associated protein that interacts with the F-actin-binding protein, cortactin. HAX-1 has been recently identified as a binding partner for canalicular Bsep, Mdr1, and Mdr2.104 There is evidence that HAX-1 and cortactin participate in clathrin-mediated Bsep endocytosis from the canalicular plasma membrane,126 and that cortactin location in the cell cortex depends on an intact actin cytoskeleton.104 Again, further elucidation of the process regulating the interplay between canalicular transporters and actin-associated proteins should be directed to understand the mechanisms underlying the canalicular-transporter internalization under redox-imbalance conditions.
Finally, mechanisms other than actin disorganization should also be considered, and may well act in concert. Examples are the changes in cell volume due to oxidative stress. Loss of hepatocellular volume and retrieval of Mrp2 from the canalicular membrane occur concomitantly after addition of tBOOH71,127 or chloro-2,4-dinitrobenzene71 to isolated perfused rat livers, and hepatocyte shrinkage induced by hyperosmotic exposure in this experimental model triggers a similar retrieval.128 However, under these hyperosmotic conditions, distribution of the tight-junctional-associated protein, ZO-1, remains unaffected.128
3.3. Signal transduction pathways and oxidative stress-induced hepatocanalicular dysfunction: «The Ca2+-PKC Connection»Elevation of cytosolic Ca2+ levels occurs under oxidative stress conditions. This is thought to involve both the entry of extracellular Ca2+ via plasma membrane receptor operated Ca2+ channels and the release of Ca2+ from intracellular Ca2+ storages, particularly in the endoplasmicreticulum (calciosome).129 Cytosolic Ca2+ elevation is a major determinant of the oxidative injury.130 As stated above, Ca2+ facilitates mitochondrial permeability transition, a key event leading to both necrosis and apoptosis. Besides, Ca2+ activates Ca2+-dependent proteases, endonucleases, and phospholipases, which reinforces mitochondrial mechanisms of cell death.11,15 However, more subtle changes in hepatocanalicular function can occur at lower, non-necrotic levels of oxidative stress, and Ca2+ elevations also seem to play a key role. The intracellular Ca2+ chelator, BAPTA/AM, fully prevented the impairment induced by low levels of tBOOH in the capability of the hepatocyte couplets to accumulate apically and retain in their canalicular vacuoles the bile salt-fluorescent analogue, CLF.110,111 Suggestively, the actin-cytoskeletal disarrangement is also prevented by this Ca2+-sequestering agent, further supporting a causal relationship between both phenomena. Furthermore, Ca2+-elevating agents, such as the Ca2+ ionophore A23187110 or the inhibitor of endoplasmic reticulum Ca2+-ATPase thapsigargin,70 mimic the deleterious effects of oxidative stress on both actin-cytoskeleton integrity and hepatocanalicular function. Taken together, these findings suggest that ROS-evoked Ca2+ signalling rather than a direct oxidation of actin plays a role in the secretory failure under mild oxidative-stress conditions.
Which are the signal pathways downstream of Ca2+ involved? Among the many ones regulating cell function, activation of Ca2+-dependent, «classical» protein kinase C isoforms (cPKCs) seems to be the more important. cPKCs contain one Ca2+-binding domain and, when cytosolic Ca2+ levels increase, recruitment of cPKCs to the plasma membrane takes place; this induces conformational changes of the enzyme leading to its activation.131 cPKC activation is a common feature under oxidative stress conditions.132 In particular, our group demonstrated that the pro-oxidant agent, tBOOH, induces cytosolic Ca2+ elevations and translocation of the cPKC isoform, PKCα, from the cytosol to the plasma membrane in isolated hepatocytes, even at concentrations low enough to only affect the biliary-secretory machinery.98 Furthermore, several previous findings showed strong similarities between the effect of oxidative stress and those induced by Ca2+ and PKC agonists, namely: i) Cytosolic Ca2+ elevations133 and PKC activation134 impaired bileflow generation in the isolated perfused rat liver,134 in part by increasing paracellular permeability;135-137 we and others further characterized these effects in the hepatocyte couplet model by showing that cytosolic Ca2+ elevations impair the couplet capability to secrete and retain in their canalicular vacuoles fluorescent bile salt analogues by activating cPKC,138 and that PKC activation by vasopressin and phorbol esters reproduced these effects.108,139,140ii) PKC agonists induce F-actin cytoskeletal disarrangements,139 and Ca2+-elevating agents reproduce these effects by a PKC-dependent mechanism.138
Final confirmation of a crucial role for cPKC activation in actin disorganization and the further impairment of hepatocanalicular function induced by ROS was provided by recent studies in hepatocyte couplets. ROS-mediated actin-cytoskeleton disarrangements were fully prevented by both PKC-pan-specific and cPKC-specific inhibitors.98 What is more relevant from the therapeutic point of view, both cytoskeleton disruption and canalicular dysfunction are reversed within 1 h by these inhibitors.98
The retrieval of the bile salt transporter, Bsep, from the canalicular membrane was also fully prevented by PKC antagonists.109 The same holds true for the impairment of the tight-junctional-retentive properties, another possible consequence of the actin disassembly induced by exposure to pro-oxidant agents.98 The exact mechanisms that explain the harmful effect of PKC on actin integrity and, by extension, to the hepatocanalicular function as a whole are unclear. Actin is a major target for PKC. This kinase phosphorylates and/or induces disorganization of a wide range of actin-cytoskeletal components, including actin itself, actin-associated proteins (e.g., α-actinin, vinculin and filamin), and membrane-cytoskeletal cross-linking proteins, including Rac (a member of the Rho family of small GTP-binding proteins), and myristoylated alaninerich C-kinase substrate (MARCKS).141,142 On the contrary, a direct phosphorylation of tight-junctional components mediated by cPKC is unlikely, since only «atypical» (non-Ca2+/diacylglycerol-dependent) PKC isoenzymes have been reported to concentrate at the tight-junctional complex.143 Alternatively, oxidative-stress-induced cPKC-dependent phosphorylation of components others than actin or actin-associated proteins is likely to contribute to Bsep retrieval. Selective activation of Ca2+-dependent PKCs by thymeleatoxin induces cholestasis and retrieval of Bsep from the canalicular membrane in the isolated perfused rat liver.144 Interestingly, the canalicular ATP-binding cassette transporter, multidrug resistance protein 1 (Mdr1), the closest homologue of Bsep, is phosphorylated by PKC at 3 serine residues at the C-terminal, «linker» region, which binds to the actin-associated protein, HAX-1.145 Whether this phosphorylation occurs also for Bsep remains to be ascertained.
The kind of canalicular protein that is internalized under oxidative-stress conditions and the signalling molecule involved seem to depends on the pro-oxidant agent employed, and on the magnitude of the oxidative damage. Low concentrations of the oxidizing compound, ethacrynic acid, does not translocate cPKC but «novel» PKC isoforms (nPKC). Under these conditions, the compound internalizes selectively Mrp2 without affecting Bsep, by a mechanism probably involving Ca2+-depending activation of inducible nitric oxide (NO) synthase (iNOS), followed by NO-mediated cGMP increase, and further cGMP-activated nPKC.113 However, higher ethacrynic acid doses, which can activate cPKC isoforms as well, induce internalization of both Bsep and Mrp2.113
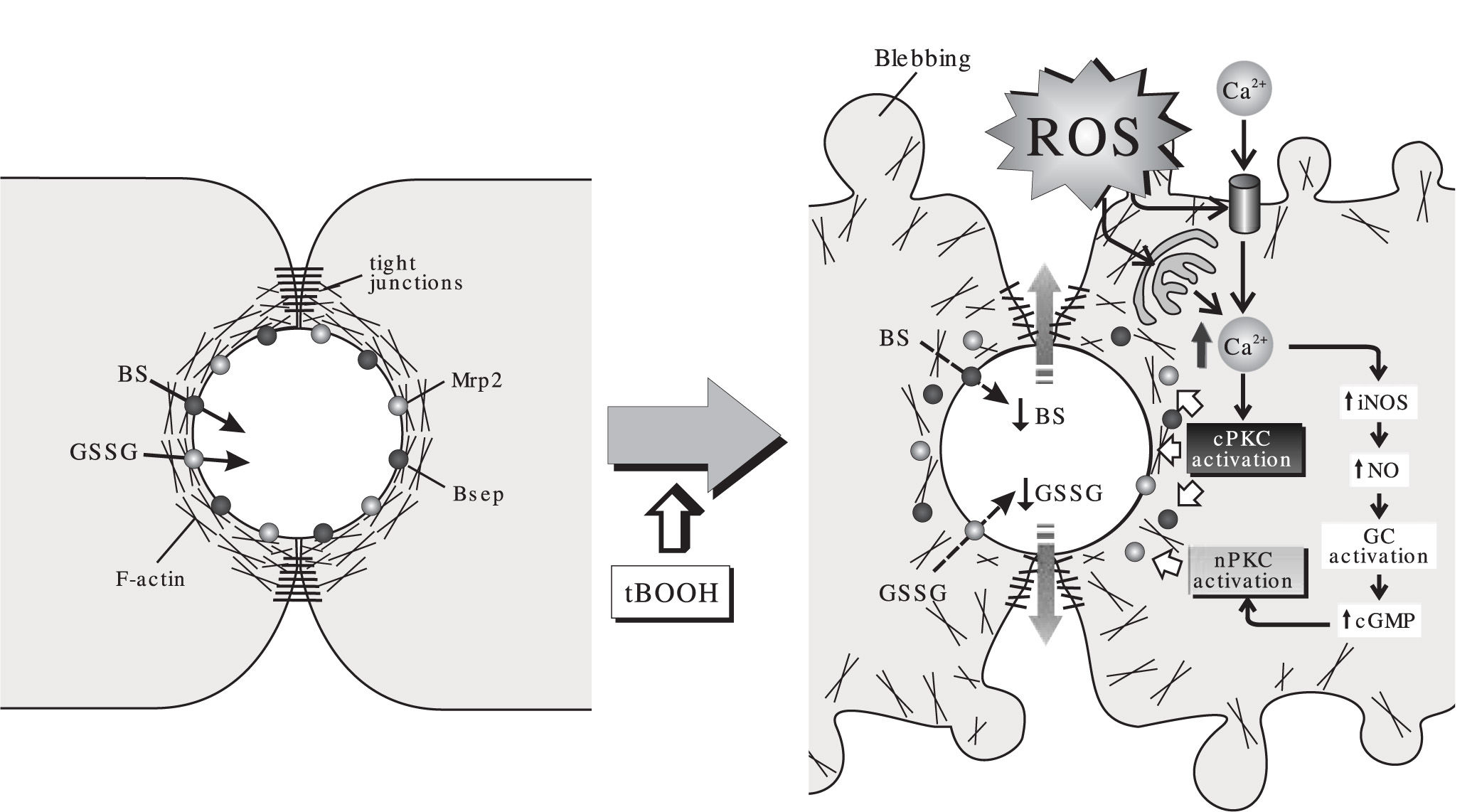
As summarized in Figure 2, a picture is emerging to understand the effect of acutely-induced oxidative stress on the hepatobiliary function. Under mild ROS challenge not affecting hepatocellular viability, Ca2+ elevations induce cPKC and/or nPKC activation. This brings on a number of alterations in both function and localization of structures relevant to bile formation, such as actin cytoskeleton, canalicular transporters and tight-junctional components. This impairs, in turn, the biliary secretion and the further retention of solutes that provide osmotic driving force for bile formation. Other factors such as GSSG-induced cis-inhibition of the bile salt transport, reduced biliary excretion of glutathione due to intracellular glutathione depletion and hepatocellular death may become contributing factors depending on the magnitude of the pro-oxidant condition.
, on the canalicular transport of monovalent bile salts (BS), excreted via the bile salt export pump (Bsep), and that of the multidrug-resistance associated protein 2 (Mrp2) substrate, oxidized glutathione (GSSG). In normal cells, the pericanalicular arrangement of the F-actin cytoskeleton allows for the appropriate insertion of the canalicular transporters in their membrane domain, and the proper barrier function of the tight-junctional structures. The acute administration of the oxidizing compound, tBOOH, stimulates formation of reactive oxygen species (ROS). This induces mobilization of Ca2+ across both the plasma and the calciosome membranes, and the subsequent activation of Ca2+-dependent («classical») PKC isoforms (cPKC). cPKC activation induces blebbing and redistribution of F-actin from the pericanalicular region to the cell body. This rearrangement, in turn, may lead to Bsep internalization and tight-junctional disorganization, which impairs BS/GSSG canalicular excretion, and their further retention inside the canalicular space. Ca2+ elevation may also activate inducible nitric oxide (NO) synthase (iNOS), which leads to NO-mediated guanylate cyclase (GC) activation and further cyclic guanosine monophosphate (cGMP)-mediated activation of «novel» PKC isoforms (nPKC). nPKC activation internalizes selectively Mrp2.")
Effect of the model oxidizing compound, tert-butylhydroperoxide (tBOOH), on the canalicular transport of monovalent bile salts (BS), excreted via the bile salt export pump (Bsep), and that of the multidrug-resistance associated protein 2 (Mrp2) substrate, oxidized glutathione (GSSG). In normal cells, the pericanalicular arrangement of the F-actin cytoskeleton allows for the appropriate insertion of the canalicular transporters in their membrane domain, and the proper barrier function of the tight-junctional structures. The acute administration of the oxidizing compound, tBOOH, stimulates formation of reactive oxygen species (ROS). This induces mobilization of Ca2+ across both the plasma and the calciosome membranes, and the subsequent activation of Ca2+-dependent («classical») PKC isoforms (cPKC). cPKC activation induces blebbing and redistribution of F-actin from the pericanalicular region to the cell body. This rearrangement, in turn, may lead to Bsep internalization and tight-junctional disorganization, which impairs BS/GSSG canalicular excretion, and their further retention inside the canalicular space. Ca2+ elevation may also activate inducible nitric oxide (NO) synthase (iNOS), which leads to NO-mediated guanylate cyclase (GC) activation and further cyclic guanosine monophosphate (cGMP)-mediated activation of «novel» PKC isoforms (nPKC). nPKC activation internalizes selectively Mrp2.
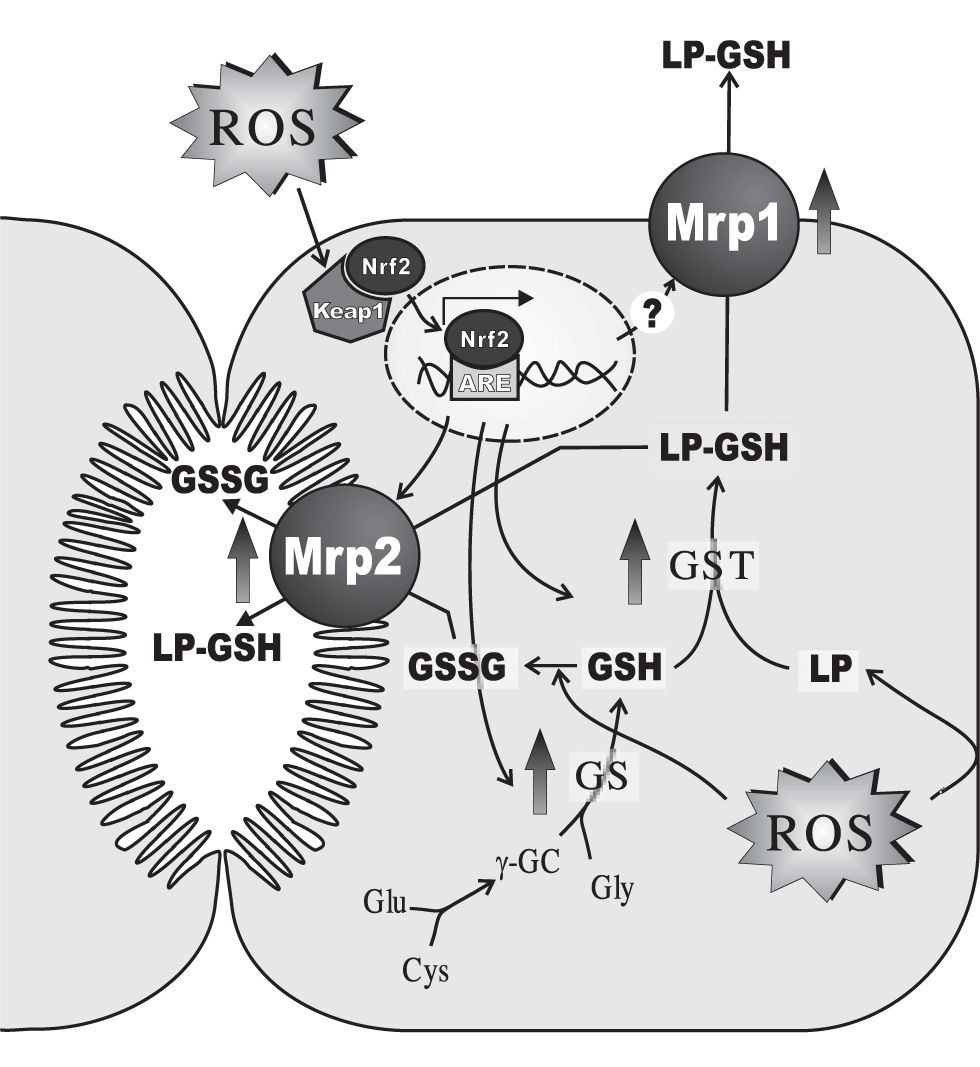
In addition to the above-mentioned alterations induced acutely following the prooxidant insult, hepatocytes develop an adaptive response against oxidative stress, when the oxidative insult is maintained with time (Figure 3). This response involves induction of antioxidant defences such as catalase146 and manganese superoxide dismutase,147 a widespread phenomenon shared with other tissues. In addition, hepatic adaptation integrates its distinctive metabolizing and secretory capacity to reinforce this adaptive defence against the oxidative insult. Glutathione synthesis is increased by induction of γ-glutamyl-cysteinyl synthetase,148 and so are the phase-II-detoxifying enzymes, GST149 and UDP-glucuronosyltransferase.147 GST induction enhances the co-ordinated inactivation, via GSH conjugation, of DNA hydroperoxides and lipid hydroperoxides formed as secondary metabolites during oxidative stress.150 GST also catalyzes GSH conjugation of highly-reactive, toxic αβ-unsaturated lipid aldehydes generated by β-scission of lipid hydroperoxides of ω-6 polyunsaturated fatty acids, 4-hydroxy trans-2-nonenal (HNE) being the most abundant.151 These lipid-peroxidation products combine spontaneously with cysteine, histidine and lysine residues of proteins, potentially modifying protein function and resulting in cellular toxicity, and have been suggested to mediate and amplify the cellular effects of their free-radical precursors.152 UDP-glucuronosyltransferase induction would improve the metabolisation, via glucuronidation, of pro-oxidant toxicants, such as benzo(a)pyrene,153 pentachlorophenol,154 acetaminophen,155 aliphatic alcohols156 and manadione.157 Phase-II products are then extruded from the cell via the hepatocellular efflux pumps, MRP1, MRP2, MRP3, MRP4 (ABCC4) and the breast cancer resistance protein (BCRP, ABCG2), all of which are also upregulated.158-160 Glutathione conjugates are substrates of MRP2 and MRP1.161,162 Since these transporters also transfer GSSG, MRP1/2-mediated GSSG extrusion helps to maintain low intracellular GSSG levels, when GSSG reduction back to GSH via GSSG reductase becomes rate-limiting. Unlike MRP1, the basolateral carriers, MRP3, MRP4 and BCRP, transport non-glutathione conjugates, including glucurono-and sulpho-conjugates, and amidated bile salts.161,162 MRP1, MRP3 and MRP4 are normally expressed at very low levels in the basolateral membrane of the hepatocytes.161,162 Upregulation of basolateral extrusion pumps during a sustained oxidant insult is expected to shift the transfer of substrates normally excreted into bile towards blood, so that to permit urinary excretion. As an untoward effect of this adaptive response, this phenomenon might decrease the biliary excretion of bile salts, thus resulting in cholestasis. The extent at which this pathomechanism contributes to the cholestasis observed under sustained oxidative stress conditions (see bellow) remains to be ascertained.
 glutathione (GSH) synthesis via GSH synthetase (GS), ii) GSH-conjugation of lipid-peroxides or their aldehydic derivatives (LP) via glutathione 5-transferase (GST), and iii) plasmatic and biliary exportation of conjugated LPs and oxidized glutathione (GSSG) via both basolateral multidrug resistance-associated protein 1 (Mrpl) and canalicular multidrug resistance-associated protein 2 (Mrp2). When cells are exposed to oxidative stress, the transcription factor Nrf2, which is bound to the negative regulator Keapl in cytosol, escapes from its repression, and translocates and accumulates in the nucleus. Once there, it binds to the antioxidant response element (ARE) and activates ARE-dependent gene expression. Proteins that are induced by Nrf2 activation include γ-glutamyl-cysteinyl synthetase (γ-GCS), GST and Mrp2. Mrpl is induced by oxidative stress by a mechanism that seems to be independent of Nrf2. Only changes in rodent-gene products are displayed, since these regulations were mostly demonstrated in this species. For further details, see Section 4.")
Main hepatocellular adaptive changes induced by sustained oxidative stress in the expression of enzymes and transporters involved in i) glutathione (GSH) synthesis via GSH synthetase (GS), ii) GSH-conjugation of lipid-peroxides or their aldehydic derivatives (LP) via glutathione 5-transferase (GST), and iii) plasmatic and biliary exportation of conjugated LPs and oxidized glutathione (GSSG) via both basolateral multidrug resistance-associated protein 1 (Mrpl) and canalicular multidrug resistance-associated protein 2 (Mrp2). When cells are exposed to oxidative stress, the transcription factor Nrf2, which is bound to the negative regulator Keapl in cytosol, escapes from its repression, and translocates and accumulates in the nucleus. Once there, it binds to the antioxidant response element (ARE) and activates ARE-dependent gene expression. Proteins that are induced by Nrf2 activation include γ-glutamyl-cysteinyl synthetase (γ-GCS), GST and Mrp2. Mrpl is induced by oxidative stress by a mechanism that seems to be independent of Nrf2. Only changes in rodent-gene products are displayed, since these regulations were mostly demonstrated in this species. For further details, see Section 4.
All these adaptive mechanisms are transcriptional in nature, and involve the coordinated activation of a number of redox-sensitive transcription factors. Early evidence that oxidative stress induces gene regulation came from the laboratories of Crawford and Cerutti,163 who reported on the induction of mRNA of the mouse protooncogenes, c-fos and c-myc, in JB6 cells by O2•— and H2O2. Soon later, a number of transcription factors, including Nrf2, NF-kB and AP-1, have been identified to be regulated by ROS, and to mediate expression of several gene products. Depending on the level of ROS, different redox-sensitive transcription factors are activated, and coordinate distinct biological responses. Low oxidative stress levels induce Nrf2,164 whereas intermediate levels of ROS trigger an inflammatory response through the activation of NF-kB and AP-1. High levels of oxidative stress surpass the defence capability of the antioxidant response, and induce mitochondrial permeability transition pores and disruption of the electron transfer, thereby resulting in apoptosis or necrosis.11
Nrf2 is a key transcription factor of the hepatic adaptation to sustained oxidative stress. Its induction has been linked to different oxidant agents such as the cancer chemoprotective agent, 3H-1,2-dimethiole-3-thione,147 alcohol,165 teributylhydroquinone,160 acetaminophen159 and bile salts.166 Activation of Nrf2 leads to the transcription of genes that codify phase II enzymes and hepatic transporters. Nrf2 induces the transcription of γ- glutamyl-cysteine synthetase,147 GST,147,149 UDPglucuronosyltransferase147,149,167 and the transporters, MRP2,158 MRP3,159 MRP4159 and BCRP;160 these upregulations were confirmed, in most cases, at the protein level. On the other hand, the role of Nrf2 in the increased expression of MRP1 at the protein level is not clear. In HepG2, induction is independent on this nuclear factor,160 whereas in knock-out mouse, the dependency is tissue-specific. For example, Nrf2 is required in fibroblast,168 but not in liver.159 These metabolic enzymes and carriers are also involved in the hepatic handling of electrophilic species that alkylate nucleophilic sites on peptides, proteins and nucleic acids, forming covalent adducts that can significantly compromise cellular integrity and function. Hence, Nrf2 is consider to be a sensor of, and a protector, against the so called «electrophilic stress».169,170
The action of Nrf2 depends on its accumulation in the nucleus, where it interacts with the antioxidant response element (ARE).171 This is a cis-acting enhancer sequence that contains the 5’-TGAC-3’ tetranucleotide present in the genes of enzymes associated with glutathione biosynthesis, redox proteins with active sulfhydryl moieties, drug-metabolizing enzymes and transporters. Nrf2 is a bZIP protein, and needs to form dimers in order to bind to ARE. It is not clear yet whether Nrf2 can interact with ARE as homodimer or it is obligated to form heterodimeric complexes with other bZIP proteins, like small RAF proteins.
The activation of Nrf2 has been suggested to be mediated by mechanisms that lead to its stabilization, increasing levels of cellular Nrf2 and subsequent transcriptional activity.172,173 Under basal conditions, Nrf2-dependent transcription is repressed by the negative regulator, Keap1. When cells are exposed to oxidative stress, electrophiles or chemopreventive agents, Nrf2 escapes Keap1-mediated repression, and translocates and accumulates in the nucleus, thus activating ARE-dependent gene expression to maintain cellular redox homeostasis.174,175 Several mechanisms have been proposed to explain the repressive effect of Keap1 on Nrf2, namely: i) Keap1 is a thiol-rich sensor protein, which is an adaptor protein for Cullin 3-dependent ubiquitination and degradation of Nrf2;176 electrophile modification of the Keap1 central linker domain would switch ubiquitination from Nrf2 to Keap1, leading to Nrf2 activation. ii) Upregulation of Nrf2 gene expression at the transcriptional level in response to ARE inducers.177iii) activation of PKC by oxidative stress and electrophiles which, in turn, activates Nrf2 through the phosphorylation of a serine residue located in the N-terminal region.178,179
5. Effect of chronic oxidative stress on hepatobiliary dysfunction: «The fight continues»The adaptive, spontaneous mechanisms that take place in hepatocytes to minimize the deleterious effects of ROS is, however, not always sufficient to prevent hepatocellular oxidative damage. Under conditions in which ROS production is maintained at high levels during long periods, alterations in the capability of the hepatocyte to produce bile and to secrete cholephilic compounds have been described in the literature. However, the information is scarce, and cannot be unequivocally ascribed to the direct effect of ROS. Some of these changes may be secondary consequences of the oxidativestress-dependent inflammatory response that develops with time under long-standing pro-oxidant conditions, the hepatic accumulation of toxic biliary solutes due to the secretory failure (e.g. bile salts, bilirubin), or unspecific effects of the agents employed to cause the pro-oxidant injury.
The Long-Evans Cinnamon rat, which mimics Wilson’s disease, is a prototypical model of chronic hepatic oxidative stress. These rats exhibit inability to mobilize copper from the liver due to a genetic defect in the Atp7b gene, which encodes for a copper-transporting P-type ATPase. This transporter localizes to the trans-Golgi network or late endosomes under basal conditions, but it relocalizes in a copper-dependent manner to a subapical, vesicular compartment, which facilitates both copper incorporation into ceruloplasmin and biliary copper excretion via lysosomes.180 Apart from copper, Long-Evans Cinnamon rats also have abnormal hepatic accumulation of iron.181 Chronic copper/iron accumulation in these rats leads to a 50% increase in lipid peroxidation, presumably due to copper/iron capability to induce oxidative stress by generating ROS via both mitochondrial dysfunction182 and Fenton-type, copper/iron-catalyzed Haber-Weiss reaction.183,184 These mutant rats exhibit a reduced basal bile salt-biliary excretion compared with normal, Long-Evans Agouti rats. This alteration is linked to a low protein expression of the canalicular bile salt transporter, Bsep.185 A posttranscriptional mechanism seems to be involved, since no changes in the levels of Bsep mRNA has been observed.186 On the other hand, mRNA levels of the bile saltuptake systems, Ntcp and Oatps (Oatp1a1 and Oatp1a4 isoforms), are decreased, although this has not been confirmed at the protein level.186 Apart from alterations in bile salt hepatic handling, Long-Evans Cinnamon rats have hyperbilirubinaemia,184,187 and impairment of the excretion of the Mrp2 substrate, BSP.188 If these alterations involve changes in the expression of Mrp2, the rate-limiting step of both bilirubin and BSP hepatic handling, this would be nontranscriptional, since Mrp2 mRNA levels are normal in these rats.186 Alternatively, the above-mentioned transcriptional downregulation of Oatps, which also transport nonbile salt organic anions such as BSP and bilirubin, may be a contributing factor. Overall, these transcriptional and post-transcriptional alterations in transporter expression may help to explain why these rats have histological features of cholestasis.187
Another experimental model of metal-induced chronic oxidative stress is that afforded by long-term aluminum (Al3+) exposure to rats. Despite Al3+ is a non-redoxcation, it bears pro-oxidant activity. Al3+ facilitates O2•— formation induced by a number of pro-oxidant maneuvers; formation of the Al3+ superoxide radical ion, AlO2•2+, has been proposed to account for this property.189 This metal also facilitates iron-driven biological oxidative reactions.190 Finally, Al3+ induces mitochondrial permeability transition, which leads to ROS formation from mitochondrial origin.191 When administered intravenously for 1-2 weeks, Al3+ reduces bile flow, and this impairment correlates directly with Al3+ hepatic content; this was associated with elevations of serum bile salts, suggesting impaired hepatic handling of bile salts.192 A more chronic exposure to Al3+ (3 months), which doubles the lipid-peroxidation levels, also reduced bile flow and the biliary output of bile salts.90 Compartmental analysis of the plasma decay of BSP revealed that both sinusoidal uptake and canalicular excretion are decreased.90 The latter phenomenon was associated with a decrease in Mrp2 protein expression. All these alterations were prevented by administration of the antioxidant, vitamin E, suggesting that oxidative stress was the main, if not the only mechanism.89
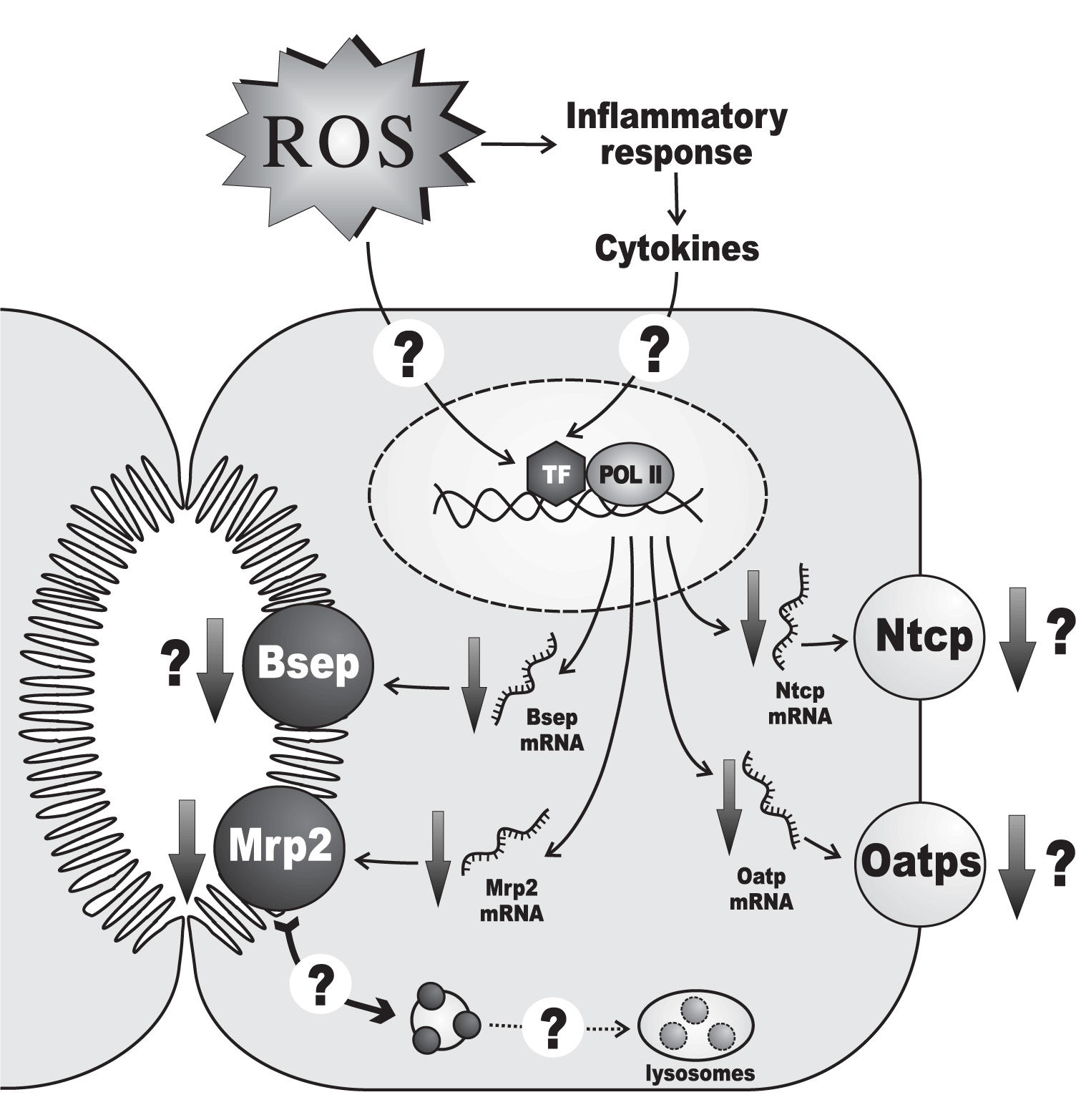
Hepatic ischemia-reperfusion injury is another prototypical oxidative-stress mediated hepatopathy associated with both cholestasis21,87 and downregulation of hepatocellular transporters193,194(Figure 4). During the ischemic phase, the hepatic cells become more susceptible to oxygen toxicity during the reperfusion phase. This is due to multiple, self-aggravating mechanisms, mainly: i) mitochondrial permeability transition occurring by cytosolic Ca2+ elevations during the ischemic phase, which induces uncoupling of electrons from the transport chain when electron flow is restored by oxygen re-entry;195-197ii) proteolytic conversion of the enzyme, xanthine dehydrogenase, to the O2•—-generating enzyme, xanthine oxidase, by proteases released during the ischemic phase upon mitochondrial damage;197,198iii) cytokine-199 and complement-200 mediated activation of Kupffer cells, with further release of chemotactic and activating cytokines to neutrophils;201-203 both Kupffer cells and neutrophils can generate ROS, which further diffuse into hepatocytes.204,205
, as well as those of the canalicular export transporters Mrp2 and Bsep. These transporter alterations have been confirmed at the protein level only for Mrp2. It is unknown whether ROS exert their effects at the transcriptional levels by directly activating certain redox-sensitive transcriptional factors (TF) or, indirectly, by promoting the release of cytokines as part of the inflammatory response secondary to the oxidative tissue injury which may, in turn, activate a different set of TFs. Since down-regulation of Mrp2 protein is more severe than that of mRNA, an additional post-transcriptional mechanism has been suggested, which may involve oxidative stress-induced transporter internalization, followed by delivery of the endocytosed transporters to the lysosomal compartment for degradation. Only changes in rodent-gene products are displayed, since these regulations were mostly demonstrated in this species. For further details, see Section 5.")
Alterations of the expression of transporters relevant to bile formation in hepatic injury by ischemia-reperfusion. An ischemic period of 60 minutes followed by 1 day of reperfusion decreases the mRNA levels of the basolateral transporters Ntcp and Oatp (all isoforms), as well as those of the canalicular export transporters Mrp2 and Bsep. These transporter alterations have been confirmed at the protein level only for Mrp2. It is unknown whether ROS exert their effects at the transcriptional levels by directly activating certain redox-sensitive transcriptional factors (TF) or, indirectly, by promoting the release of cytokines as part of the inflammatory response secondary to the oxidative tissue injury which may, in turn, activate a different set of TFs. Since down-regulation of Mrp2 protein is more severe than that of mRNA, an additional post-transcriptional mechanism has been suggested, which may involve oxidative stress-induced transporter internalization, followed by delivery of the endocytosed transporters to the lysosomal compartment for degradation. Only changes in rodent-gene products are displayed, since these regulations were mostly demonstrated in this species. For further details, see Section 5.
Hepatic oxidative stress as a consequence of ischemiareperfusion depends on the duration of the ischemic phase.206,207 A 30-minute ischemia followed by different reperfusion periods either-in the isolated perfused rat liver model208 or in vivo86,207,209,210 does not affect both the GSSG hepatic content and/or the hepatic lipid-peroxidation levels, measured by generation of the secondary lipid-peroxidation by-products, such as malondialdehyde and HNE. This should not been interpreted as complete absence of oxidative stress, however, since the sensitivity of the methods measuring secondary lipid-peroxidation products is limited as compared with those measuring primary lipid peroxides,207,211,212 and the enzyme, glutathione reductase, may fully account for the conversion of GSSG back to GSH at low ROS levels;213 alternatively, ROS production may be too low to account for effective GSH conversion to GSSG.214 On the contrary, longer ischemic periods (45-90 minutes) followed by reperfusion result in increased lipid peroxidation, even assessing secondary lipid-peroxidation products.87,210,215-220
The impact of this maneuver on both bile-flow generation and the expression of transporters also depends on the duration of the ischemia. A 30-minute ischemia decreases both bile flow and biliary bile salt output after 1 day of reperfusion, but no changes in mRNA and protein levels of the basolateral and canalicular main bile salt transporters, Ntcp and Bsep, respectively, have been recorded.86 Functional/localization alterations of these transporters, or impairment of the expression of other bile salt transporters, such as Oatps, may be involved in the bile salt-secretory failure. Similarly, Mrp2 mRNA and protein expressions are unaffected, in agreement with the absence of changes in the maximum secretory rate of the Mrp2 substrate, ceftriaxone;86 lack of studies of ceftriaxone excretion under non-saturating conditions, however, does not allow changes in intrinsic transport efficiency to be ruled out. Unlike a 30-minute ischemia, a 60-minute ischemic period followed by 1 day of reperfusion decreases the mRNA levels of the basolateral transporters, Ntcp and Oatp (all isoforms), as well as those of the canalicular export transporters, Mrp2 and Bsep.193 This has been confirmed at the protein level for Mrp2.194 At least part of these transcriptional alterations may be due to the ROS-dependent hepatic inflammatory response, which leads to release of cholestatic, proinflammatory cytokines with capability to impair transporter expression at a transcriptional level.221 However, down-regulation of Mrp2 protein is more profound than that of mRNA, suggesting additional post-transcriptional mechanisms.194 Although the causes underlying the latter phenomenon are presently unknown, the early oxidative stress-induced transporter internalization, sustained with time, may lead to the delivery of the endocytosed transporters to the lysosomal compartment, followed by degradation, as was suggested to occur late in lipopolysaccharide-induced cholestasis222 and in obstructive cholestasis223 in rats. Apart from alterations in transporter expression/function, the tight-junctional barrier is impaired in ischemia-reperfusion injury, as shown in 24-or 48-h-could-stored isolated perfused rat livers subjected to reperfusion.224
Taken together, these models of long-lasting oxidative stress show consistently that cholestasis and/or impairment of the constitutive expression of transporters relevant to bile formation is a common feature in prolonged oxidative stress, and that both transcriptional and posttranscriptional mechanisms are involved. Proinflammatory cytokines released by the inflammatory response to the oxidative liver damage may be key mediators of part of these alterations.
6. Concluding remarks and future directions: «The end… of the beginning»Oxidative stress is a cholestatic condition. Three consecutive phases in the oxidative injury affecting biliary secretion can be distinguished. Initially, the oxidative insult impairs acutely the hepatobiliary function, even at oxidative stress levels far lower than those affecting hepatocellular viability. These alterations comprise early impairment in hepatocyte capability to secrete and retain in the bile canaliculus osmotically-active solutes that are main determinants of bile generation, such as bile salts and GSH/GSSG. A key role for actin-cytoskeletal disarrangement on these functional alterations has been proposed, since it has been causally linked to internalization of canalicular transporters and disarrangement of tight-junctional structures. ROS-evoked, Ca2+-dependent signalling events, such as cPKC and/or nPKC activation, seem to mediate these harmful effects.
To limit these primary, deleterious consequences, hepatocytes develop a secondary adaptive response, triggered by ROS-responsive transcription factors, mainly the Nfr2/Keap1 system. This protective response involves up-regulations of the GSH-synthesizing enzyme, GSH-synthetase, the GSH-conjugating enzyme, GST, and the hepatocellular efflux pumps, Mrp2 and Mrp1. This enhances the co-ordinated inactivation of DNA/lipid hydroperoxides and their derivatives via GST-mediated GSH conjugation, and their further cellular extrusion via Mrp2 and Mrp1. Since, this adaptive response also involves upregulation of other basolateral pumps that are engaged in the transport of bile salts (Mrp3 and Mrp4), this may represent an additional mechanism of cholestasis, by shifting the transfer of these osmotically-active compounds from bile to blood.
Finally, a prolonged/high oxidative-stress challenge surpassing this adaptive antioxidant defence, such as that induced by ischemia-reperfusion or by accumulation of metals (e.g., Cu2+, Al3+), induces a number of alterations in hepatocellular transporters both at functional and expression levels. For the latter case, both transcriptional and posttranscriptional mechanisms have been reported, and the inflammatory response secondary to the oxidative tissue injury may play a key role.
Despite considerable progresses have been made in the characterization of the effects of oxidative stress on the biliary secretory machinery, the characterization of the molecular mechanisms underlying these effects is in its infancy. A bridge needs to be built between early events and late consequences of oxidative stress on bile secretion, in order to reconstruct the cascade of events leading to the posttranscriptional changes in transporter protein expression observed eventually during the sustained oxidative challenge. Also, we need to distinguish direct ROS-mediated effects from secondary consequences of the oxidative injury (e.g., inflammatory response, accumulation of biliary solutes). In addition, we must fully characterize the redox-sensitive signalling pathways involved in these effects; a more complete picture should provide more selective therapeutic strategies to interfere with ROS-mediated harmful pathways, or to enhance the protective ones. Studies on the impact of oxidative stress in transport function of biliary epithelial cells are also eagerly awaited; cholangiocytes may contribute to bile secretory failure, as they are the main target of cholangiopathies associated with periductal inflammation and oxidative stress. And last, but not least, it remains to be ascertained the actual contribution of oxidative stress in both transcriptional and posttranscriptional changes in liver transporter expression occurring in chronic cholestatic liver diseases in humans.161
Genomic and proteomic tools are accelerating the discovery of new ROS-responsive genes and the molecular targets of ROS action. These approaches are expected to greatly help to achieve the goals above. Meanwhile, we hope this preliminary information contributes to draw attention about the convenience of limiting oxidative stress in hepatopathies. Some short-scale clinical studies using co-adjuvant, antioxidant therapies have shown encouraging results,22 but multicentric and long-term clinical trials are needed to determine whether this strategy holds promise for the future.
Acknowledgements.This work was supported by CONICET (PIP 6442) and Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT; PICT 0526115), and Fundación Florencio Fiorini, Argentina.



