



Necroptosis and endoplasmic reticulum (ER) stress has been implicated in acute and chronic liver injury. Activated eukaryotic initiation factor 2 alpha (eIF2α) attenuates protein synthesis and relieves the load of protein folding in the ER. In this study, we aimed to analyze the impact of eIF2α phosphorylation on hepatocyte necroptosis in acute liver injury.
Materials and methodsMale BALB/c mice were injected with tunicamycin or d-galactosamine, and LO2 cells were incubated with tunicamycin to induce acute liver injury. 4-Phenylbutyric acid (PBA) and salubrinal were used to inhibit ER stress and eIF2α dephosphorylation, respectively. We analyzed the eIF2α phosphorylation, ER stress, and hepatocyte necroptosis in mice and cells model.
ResultsTunicamycin or d-galactosamine significantly induced ER stress and necroptosis, as well as eIF2α phosphorylation, in mice and LO2 cells (p<0.05). ER stress aggravated tunicamycin-induced hepatocyte necroptosis in mice and LO2 cells (p<0.05). Elevated eIF2α phosphorylation significantly mitigated hepatocyte ER stress (p<0.05) and hepatocyte necroptosis in mice (34.37±3.39% vs 22.53±2.18%; p<0.05) and LO2 cells (1±0.11 vs 0.33±0.05; p<0.05). Interestingly, tumor necrosis factor receptor (TNFR) 1 protein levels were not completely synchronized with necroptosis. TNFR1 expression was reduced in d-galactosamine-treated mice (p<0.05) and cells incubated with tunicamycin for 12 and 24h (p<0.05). ER stress partially restored TNFR1 expression and increased necroptosis in tunicamycin-incubated cells (p<0.05).
ConclusionsThese results imply that ER stress can mediate hepatocyte necroptosis independent of TNFR1 signaling and elevated eIF2α phosphorylation can mitigate ER stress during acute liver injury.
The liver is an important metabolic and detoxifying organ of the body. Due to its unique function and dual blood supply, it is often exposed to a large number of toxins and exogenous substances including alcohol, drugs, and hepatoviral infections [1]. The metabolism of these toxic substances and liver infections can cause liver injuries that can progress to end-stage liver disease. Acute and chronic liver injuries share a common mechanism that can be attributed to hepatocyte degeneration and cell death [2,3]. Cell death is mediated through different modalities, including apoptosis, autophagy, necrosis, necroptosis, and cornification [4]. Therefore, controlling hepatocyte death can be an effective strategy to improve liver injury. Necroptosis, a newly discovered form of cell death, is an inflammatory form of necrotic cell death that resembles the morphological features of necrosis, yet the process is tightly regulated like apoptosis [5]. Hepatocyte necroptosis has been implicated in the pathogenesis of various liver diseases [2,3,6,7], such as liver injury caused by nonalcoholic fatty liver disease (NAFLD) or nonalcoholic steatohepatitis (NASH), alcohol [8,9], carbon tetrachloride [10], drug-induced liver injury, hypoxia-induced liver injury [11], pyrazinamide, and paracetamol-induced liver injury [12]. Necroptotic signaling is mediated by death receptors, such as tumor necrosis factor-alpha (TNF-α), TNF receptor 1 (TNFR1), and toll-like receptors (TLR) that phosphorylates the receptor-interacting protein 3 (RIP3), which in turn recruits and phosphorylates the mixed lineage kinase domain-like pseudokinase (MLKL). Phosphorylation of MLKL increases the permeabilization of the cell membrane [13]. Therefore, RIP3 and MLKL are considered to be key modulators of necroptosis.
The endoplasmic reticulum (ER) is an organelle that plays critical roles in the synthesis and folding of proteins [14]. In addition, the ER maintains the calcium homeostasis of cells, as well as it mediates the biosynthesis of phospholipids and cholesterols. Disruption the function of the ER leads to the accumulation of unfolded or misfolded proteins in the ER lumen, which in return, induces ER stress [7]. ER stress has been shown to induce necroptosis [15,16] through activation of the pro-apoptotic signaling cascade [17]. ER stress causes the glucose-regulated protein 78 (GRP78) to competitively bind unfolded or misfolded proteins [18]. In turn, this leads to the phosphorylation of protein kinase R-like ER kinase (PERK) and inositol-requiring enzyme 1 alpha (IRE1α), along with the cleavage of activating transcription factor 6 (ATF6) [19]. Subsequently, activated PERK can phosphorylate the alpha subunit of eukaryotic initiation factor 2 (eIF2α) at serine 51 [20]. eIF2 is a 126kDa protein composed of α, β and γ subunits that are essential for mRNA translation and subsequent protein synthesis [21]. The phosphorylated eIF2α attenuates protein synthesis and thus relieves the protein folding load in the ER [22]. Simultaneously, phosphorylated eIF2α activates another cascade that leads to the dephosphorylation of eIF2α, which creates a negative feedback mechanism to restore protein synthesis [23]. Briefly, phosphorylated eIF2α up-regulates the translation of activating transcription factor 4 (ATF4), which in turn induces the expression of growth arrest and DNA damage 34 (GADD34) as well as GRP78 and C/EBP homologous protein (CHOP) [24]. GADD34 binds with protein phosphatase 1 (PP1), thereby facilitating the dephosphorylation of eIF2α [25].
ER stress can also be relieved by several chemical reagents. For example, 4-phenylbutyric acid (PBA) is a chemical chaperone that has been shown to alleviate ER stress in different cell models [26–28]. Salubrinal selectively reduces the dephosphorylation of eIF2α by suppressing the activity of PP1, therefore maintaining the phosphorylated eIF2α levels and inhibiting the expression of eIF2 as and minimizing ER stress [29,30]. Furthermore, PERK, ATF6, and IRE1α can also activate ER-related degradation and promoting cell survival [31].
Tunicamycin is a nucleoside antibiotic that can be used as a pharmacological inducer of ER stress [32,33]. d-Galactosamine is a hepatotoxic agent that causes the inhibition of transcription and protein synthesis, which leads to cell death [34]. TNF-α and ER stress are involved in the pathogenesis of several liver diseases [35]. Previous studies demonstrated that tunicamycin could induce acute kidney injury by inducing ER stress [36,37]. Furthermore, tunicamycin can be used to induce acute liver injury [38]. In this study, we used tunicamycin and d-galactosamine-induced acute liver injury mouse models and tunicamycin-induced ER stress cell models to investigate the regulatory functions of eIF2α phosphorylation in hepatocyte necroptosis in acute liver injury.
2Materials and methods2.1AnimalA total of 400 male BALB/c mice (6–8 weeks old, 18±2g) were obtained from the Animal Center of Zunyi Medical College (Guizhou, China). The animals were housed in a specific pathogen-free facility with the temperature maintained between 20 and 24°C and an automatic 12-h light/dark cycle with food and water available ad libitum. The study protocol was approved by the Animal Care and Use Committee of the Affiliated Hospital of Zunyi Medical College (Guizhou Province, China) in accordance with Guidelines of China Animal Care and Research.
2.2Experimental designMice were allowed one week to acclimatize before commencing the experimental procedures. Next, the mice were randomly divided into different experimental groups. The control group mice (n=10 mice) received an intraperitoneal injection of phosphate buffer saline (PBS, 10mL/kg). To induce acute liver injury, mice either received an intraperitoneal injection of tunicamycin (2mg/kg, Sigma) in the tunicamycin group (n=10 mice) or d-galactosamine (1000mg/kg, Sigma) in the d-galactosamine-treated mice (n=10 mice). To pharmacologically alleviate the ER stress, mice were pretreated with an intraperitoneal injection of PBA (150mg/kg, Sigma) or salubrinal (1mg/kg, Sigma) for 2h. Subsequently, the mice were administered tunicamycin resulting in PBA+tunicamycin, and salubrinal+tunicamycin treatment groups (n=10 mice per group). Alternatively, the PBS, PBA, and salubrinal treated mice were given a PBS injection to serve as control and PBA or salubrinal groups, respectively (n=10 mice per group).
2.3Cell culture and ER stress inductionThe human hepatocyte LO2 cell line was obtained from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). LO2 cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. To investigate the impact of tunicamycin on ER stress and necroptosis, LO2 cells were treated with PBS or tunicamycin (1μg/mL, Sigma) for 12, 24, or 48h. Similarly, PBA and salubrinal were used to alleviate ER stress. Briefly, LO2 cells were pretreated with PBA (10mM) or salubrinal (20μM, Sigma) for 2h and then incubated with PBS (control) or tunicamycin (1μg/mL, Sigma).
2.4Western blotMice were sacrificed, and the livers were dissected and homogenized in immunoprecipitation assay lysis buffer (10mg/mL, R0010, Solarbio, Beijing, China). Following centrifugation, individual liver lysates (40μg) were separated on 10% sodium dodecyl sulfate polyacrylamide gels by electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). Membranes were blocked with 5% skim milk in TBS and probed with the following mouse monoclonal antibodies against β-actin (sc-58673, 1:10000, sc: Santa Cruz Biotechnology), CHOP (ab11419, 1:10000), eIF2α (sc-133132, 1:10000), GRP78 (sc-376768, 1:10000), phosphorylated PERK (p-PERK, MA5-15033, 1:10000, Thermo Fisher Scientific, USA), PERK (sc-377400, 1:10000), RIP3 (sc-374639, 1:10000) and TNFR1 (sc-374186, 1:10000) or rabbit monoclonal antibodies against cleaved caspase-3 (9664, 1:10000, Cell Signaling Technology) and phosphorylated eIF2α (p-eIF2α, 3398, 1:10000, Cell Signaling Technology) at 4°C overnight. Next, the bound antibodies were detected with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit IgG and visualized using enhanced chemiluminescence. The relative level of each target protein was calculated by densitometric analysis using the Quantity One software (Bio-Rad, Hercules, CA, USA).
2.5Cell viability assayCell viability was assessed using the MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] method with the Cell Titer 96 AQueous One Solution Cell Proliferation Assay kit (Promega Corporation, Madison, WI, USA) according to the manufacturer's protocol. Briefly, the LO2 cells were incubated with 20μL of MTS solution for 3h at 37°C, and the absorbance was measured at 490nm on a microplate reader (Bio-Rad model 680; Bio-Rad, Hercules, CA, United States). LO2 cell viability was normalized as a percentage of the control.
2.6Histology and immunohistochemistryLiver tissues were fixed in 10% formalin, embedded in paraffin, and sectioned at 5μm thickness on a microtome (Leica). Following deparaffinization and rehydration, the tissue sections were stained with hematoxylin and eosin (H&E) according to the standard protocols. Alternatively, the sections were blocked and subsequently immunostained using monoclonal antibodies against RIP3 (sc-374639, 1:200) and TNFR1 (sc-374186, 1:200). The signal was visualized using the DAB reagent under a light microscope.
2.7Serum alanine aminotransferase levelFollowing euthanasia, sera samples were obtained from the blood of each mouse. Serum alanine aminotransferase (ALT) levels were determined using the rate method with the Beckman Coulter Auto Analyzer (Model AU5800, USA) according to the standard protocols.
2.8Statistical analysisThe differences between the examined groups were calculated using Student's t-test. Numerical data were expressed as mean±standard deviation (SD). Survival rate was estimated using the Kaplan–Meier method and the log-rank test. A p-value <0.05 was considered to be statistically significant.
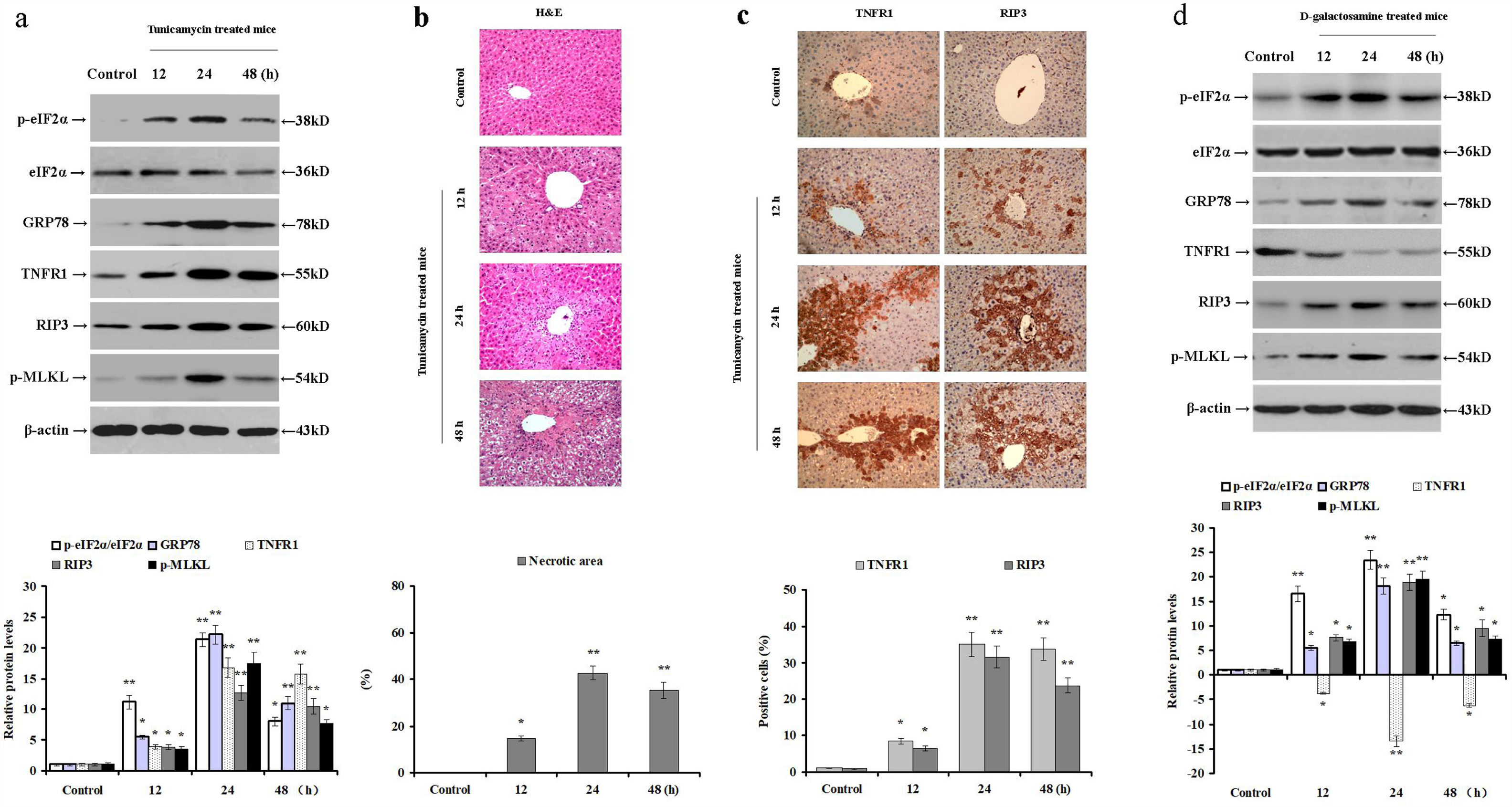
3Results3.1Tunicamycin and d-galactosamine induces ER stress and necroptosis in miceTunicamycin disrupts protein folding in the ER, resulting in ER stress and hepatocyte injury. TNF-α induces necroptosis through binding of TNFR1. Therefore, we challenged male BALB/c mice with tunicamycin or d-galactosamine injections to induce acute liver injury and analyzed the relative protein levels of intrahepatic TNFR1, p-eIF2α, eIF2α, GRP78, RIP3, and p-MLKL. In the livers of tunicamycin-treated mice, the expression of TNFR1, GRP78, and RIP3, and phosphorylated levels of eIF2α and MLKL were increased (p<0.05; Fig. 1a). Similarly, signs of hepatocyte necrosis were confirmed by H&E staining especially at 24 and 48h post-injection (p<0.05; Fig. 1b). Compared to the control group, the intrahepatic expression of TNFR1 and RIP3 were upregulated in the tunicamycin-treated mice, especially around the central vein of a hepatic lobule at 24 and 48h post tunicamycin injection (p<0.05; Fig. 1c). These results clearly demonstrate that tunicamycin administration induces ER stress, hepatocyte TNFR1 expression, eIF2α phosphorylation, and necroptosis in mice.
, tunicamycin (tunicamycin group), or d-galactosamine (d-galactosamine group). (a) The relative expression of intrahepatic TNFR1, p-eIF2α, eIF2α, GRP78, RIP3, and p-MLKL were determined by Western blot at 12, 24 and 48h post tunicamycin injection (n=10 mice per group). (b) Hepatocyte necrosis was detected by in H&E staining in paraffin-embedded sections. (c) Immunohistochemical staining of intrahepatic TNFR1 and RIP3 expression in mice (magnification 100×). (d) The relative expression of intrahepatic TNFR1, p-eIF2α, eIF2α, GRP78, RIP3, and p-MLKL was determined by Western blot at 12, 24 and 48h post d-galactosamine administration (n=10 mice per group). *p<0.05, **p<0.01 versus the control group.")
Tunicamycin or d-galactosamine administration induces ER stress and hepatocyte necroptosis in mice. Male BALB/c mice were injected with either PBS (control group), tunicamycin (tunicamycin group), or d-galactosamine (d-galactosamine group). (a) The relative expression of intrahepatic TNFR1, p-eIF2α, eIF2α, GRP78, RIP3, and p-MLKL were determined by Western blot at 12, 24 and 48h post tunicamycin injection (n=10 mice per group). (b) Hepatocyte necrosis was detected by in H&E staining in paraffin-embedded sections. (c) Immunohistochemical staining of intrahepatic TNFR1 and RIP3 expression in mice (magnification 100×). (d) The relative expression of intrahepatic TNFR1, p-eIF2α, eIF2α, GRP78, RIP3, and p-MLKL was determined by Western blot at 12, 24 and 48h post d-galactosamine administration (n=10 mice per group). *p<0.05, **p<0.01 versus the control group.
Similarly, d-galactosamine administration increased RIP3 and GRP78 expression, as well as phosphorylated eIF2α and MLKL levels in mice (p<0.05; Fig. 1d). Nevertheless, d-galactosamine treatment reduced the intrahepatic expression of TNFR1 compared to the control mice (p<0.05; Fig. 1d). These data demonstrate that d-galactosamine induces ER stress and necroptosis in mice without up-regulating hepatic TNFR1 expression. Therefore, it is plausible to speculate that hepatocyte necroptosis can be mediated by TNFR1-independent pathways.
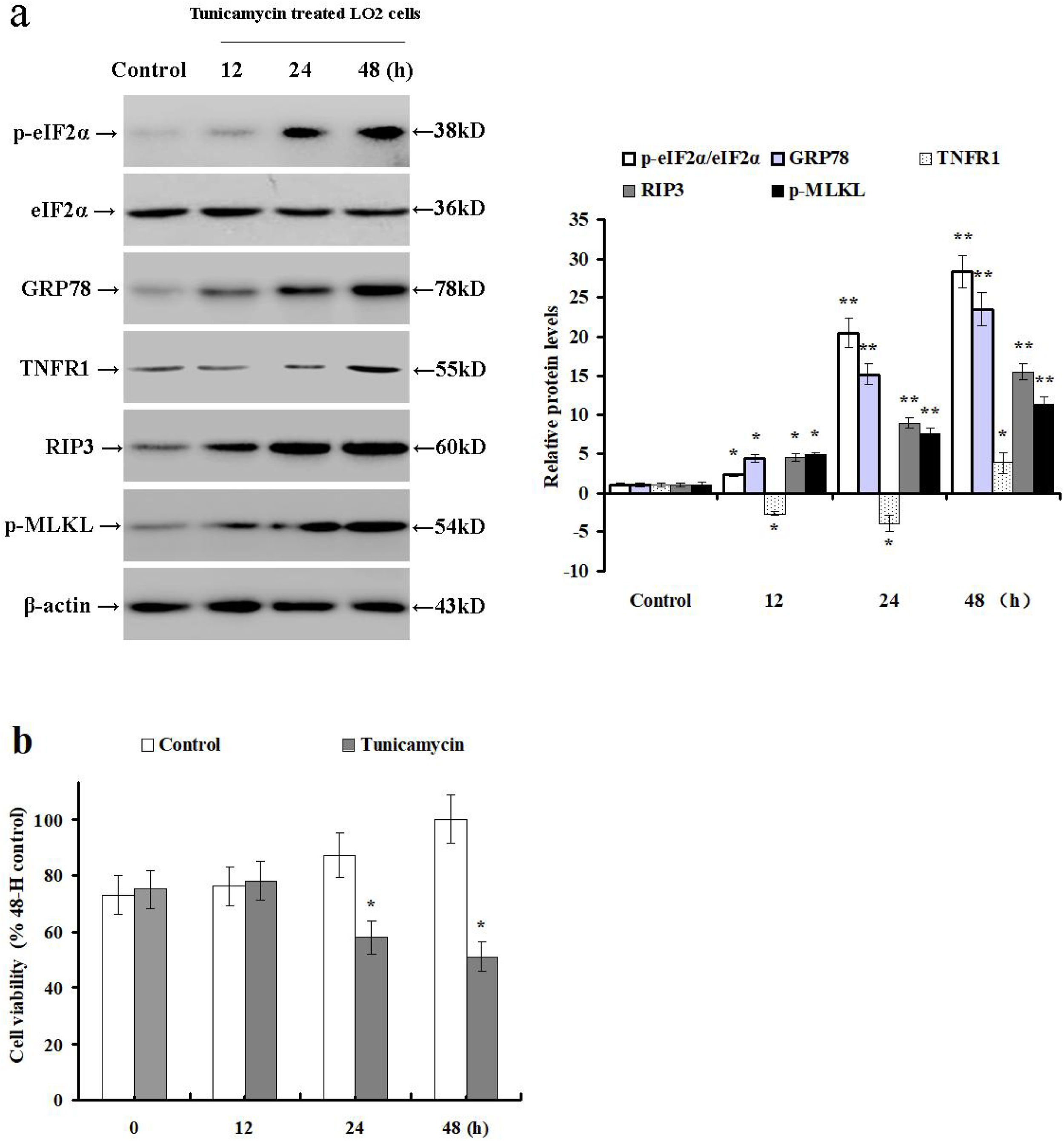
3.2Tunicamycin treatment induces ER stress and necroptosis while reducing TNFR1 expression in LO2 cellsThe results outlined above suggest that hepatocyte necroptosis can be mediated via TNF-α/TNFR1-independent pathways. Therefore, we treated LO2 cells with tunicamycin to investigate the impact of ER stress on TNFR1 expression and necroptosis. Next, we examined the relative expression of TNFR1, p-eIF2α, eIF2α, GRP78, RIP3 and p-MLKL by Western blot. Tunicamycin treatment increased GRP78 and RIP3 expression, as well as phosphorylated eIF2α and MLKL levels when compared with the control cells (p<0.05; Fig. 2a). Additionally, tunicamycin treatment significantly down-regulated the TNFR1 protein expression at 12 and 24h, then increased its expression at 48h. It also reduced the LO2 cell viability at 24 and 48h as compared to the control cells (p<0.05; Fig. 2b). These data demonstrate that tunicamycin treatment can induce ER stress and hepatocyte necroptosis via TNFR1-independent pathways in LO2 cells.
 or tunicamycin (tunicamycin group). (a) The relative expression of intrahepatic TNFR1, p-eIF2α, eIF2α, GRP78, RIP3, and p-MLKL were determined by Western blot in LO2 cells at 12, 24 and 48h post incubation with tunicamycin. (b) Viability of LO2 cells was determined by MTS assay. Histograms represent mean±SD of five independent experiments. *p<0.05, **p<0.01 versus the control group.")
Tunicamycin treatment induces hepatocyte ER stress and necroptosis in LO2 cells. LO2 cells were treated with PBS (control group) or tunicamycin (tunicamycin group). (a) The relative expression of intrahepatic TNFR1, p-eIF2α, eIF2α, GRP78, RIP3, and p-MLKL were determined by Western blot in LO2 cells at 12, 24 and 48h post incubation with tunicamycin. (b) Viability of LO2 cells was determined by MTS assay. Histograms represent mean±SD of five independent experiments. *p<0.05, **p<0.01 versus the control group.
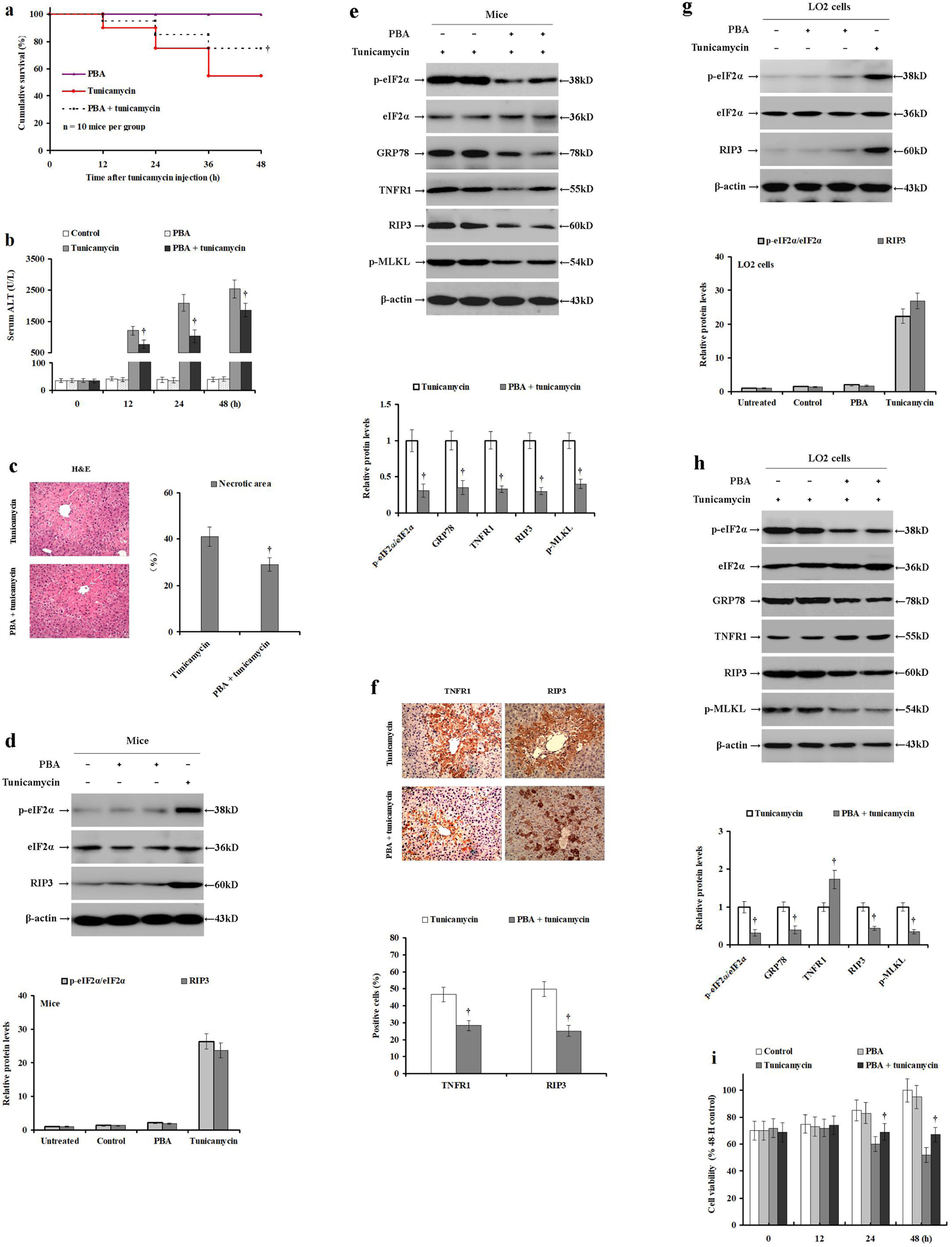
PBA alleviates ER stress, which produces hepatoprotective effects. To confirm this hypothesis, male BALB/c mice were pretreated with PBS or PBA and injected with tunicamycin or PBS. Compared with the tunicamycin-treated mice, the cumulative survival was significantly improved in mice pretreated with PBA before tunicamycin injection (p<0.05; Fig. 3a). However, PBA treatment did not significantly affect the mortality rate in the control mice (Fig. 3a). Tunicamycin treatment significantly increased serum ALT levels when compared with the control or PBA treated mice. Tunicamycin with PBA pretreatment significantly reduced the serum ALT levels in mice (p<0.05; Fig. 3b). Similarly, tunicamycin with PBA pretreatment improved the hepatic necrosis as revealed by H&E staining (p<0.05; Fig. 3c). Interestingly, PBA treatment did not significantly change eIF2α phosphorylation levels or RIP3 expression levels in mice when compared with the control group without ER stress (Fig. 3d). On the other hand, PBA pretreatment before tunicamycin significantly reduced TNFR1, RIP3 and GRP78 expression, as well as eIF2α and MLKL phosphorylation (p<0.05; Fig. 3e). Similarly, PBA pretreatment before tunicamycin decreased the expression of TNFR1 and RIP3 in the liver samples as compared to the tunicamycin-treated mice (p<0.05; Fig. 3f). These data clearly demonstrate that PBA pretreatment alleviates tunicamycin-induced liver injury, hepatocyte ER stress, necroptosis, while also down-regulating TNFR1 expression in mice.
 The cumulative survival of each mouse group was investigated at 0, 12, 24, 36 and 48h (n=10 mice per group). (b) Serum ALT levels were detected using the rate method in the control (PBS and PBS), PBA (PBA pretreatment and PBS injection), tunicamycin (PBS and tunicamycin injection), and the PBA+tunicamycin (PBA pre-treatment and tunicamycin injection) groups (n=10 mice per group). (c) Histological examination of hepatocyte necrosis in the mice livers of tunicamycin and tunicamycin/PBA pretreated group (n=10 mice per group). (d) The relative expression intrahepatic p-eIF2α, eIF2α, and RIP3 post-PBA treatment in mice compared to the control mice. (e) The relative expression of intrahepatic TNFR1, p-eIF2α, eIF2α, GRP78, RIP3, and p-MLKL were determined by Western blot in the tunicamycin group and the tunicamycin/PBA pretreated mice. (f) Immunohistochemical analysis of intrahepatic TNFR1 and RIP3 expression in the paraffin-embedded liver tissues (magnification 100×, n=10 mice per group). (g) The relative expression of p-eIF2α, eIF2α, and RIP3 were determined by Western blot in the untreated, control (PBS and PBS incubation), and tunicamycin (PBS and tunicamycin incubation) LO2 cells. (h) The relative expression of TNFR1, p-eIF2α, eIF2α, GRP78, RIP3, and p-MLKL were determined by Western blot in the tunicamycin group and the tunicamycin/PBA pretreated LO2 cells. Protein samples were electrophoresed in duplicate lanes. (i) Viability of LO2 cells as determined by MTS assay in the control (PBS and PBS incubation), PBA (PBA pretreatment and PBS incubation), tunicamycin (PBS and tunicamycin incubation), and the PBA+tunicamycin (PBA pretreatment and tunicamycin incubation) groups. Histograms represent mean±SD of four independent experiments (n=10 mice per group). †p<0.05 versus the tunicamycin group.")
PBA pretreatment moderates tunicamycin-induced ER stress and hepatocyte necroptosis in mice and LO2 cells. Male BALB/c mice were pretreated with PBS or PBA for 2h, and then injected with PBS or tunicamycin for 24h. LO2 cells were pretreated with PBS or PBA for 2h, and then injected with PBS or tunicamycin. (a) The cumulative survival of each mouse group was investigated at 0, 12, 24, 36 and 48h (n=10 mice per group). (b) Serum ALT levels were detected using the rate method in the control (PBS and PBS), PBA (PBA pretreatment and PBS injection), tunicamycin (PBS and tunicamycin injection), and the PBA+tunicamycin (PBA pre-treatment and tunicamycin injection) groups (n=10 mice per group). (c) Histological examination of hepatocyte necrosis in the mice livers of tunicamycin and tunicamycin/PBA pretreated group (n=10 mice per group). (d) The relative expression intrahepatic p-eIF2α, eIF2α, and RIP3 post-PBA treatment in mice compared to the control mice. (e) The relative expression of intrahepatic TNFR1, p-eIF2α, eIF2α, GRP78, RIP3, and p-MLKL were determined by Western blot in the tunicamycin group and the tunicamycin/PBA pretreated mice. (f) Immunohistochemical analysis of intrahepatic TNFR1 and RIP3 expression in the paraffin-embedded liver tissues (magnification 100×, n=10 mice per group). (g) The relative expression of p-eIF2α, eIF2α, and RIP3 were determined by Western blot in the untreated, control (PBS and PBS incubation), and tunicamycin (PBS and tunicamycin incubation) LO2 cells. (h) The relative expression of TNFR1, p-eIF2α, eIF2α, GRP78, RIP3, and p-MLKL were determined by Western blot in the tunicamycin group and the tunicamycin/PBA pretreated LO2 cells. Protein samples were electrophoresed in duplicate lanes. (i) Viability of LO2 cells as determined by MTS assay in the control (PBS and PBS incubation), PBA (PBA pretreatment and PBS incubation), tunicamycin (PBS and tunicamycin incubation), and the PBA+tunicamycin (PBA pretreatment and tunicamycin incubation) groups. Histograms represent mean±SD of four independent experiments (n=10 mice per group). †p<0.05 versus the tunicamycin group.
To further confirm our hypothesis, the LO2 cells were pretreated with PBS or PBA, and then incubated with tunicamycin or PBS. PBA treatment did not change eIF2α phosphorylation or RIP3 expression in LO2 cells compared to the control LO2 without ER stress (Fig. 3g). However, tunicamycin incubation increased eIF2αphosphorylation and RIP3 expression in LO2 cells. Interestingly, PBA pretreatment before tunicamycin incubation significantly reduced GRP78 and RIP3 expression, as well as eIF2α and MLKL phosphorylation (p<0.05; Fig. 3h). In contrast, it significantly restored the TNFR1 expression in LO2 cells. In addition, LO2 viability was significantly enhanced in the tunicamycin/PBA pretreated group compared to the tunicamycin group (p<0.05, Fig. 3i). These results demonstrate that PBA pretreatment can moderate tunicamycin-induced ER stress and hepatocyte necroptosis in a manner independent of TNF-α/TNFR1 signaling in LO2 cells.
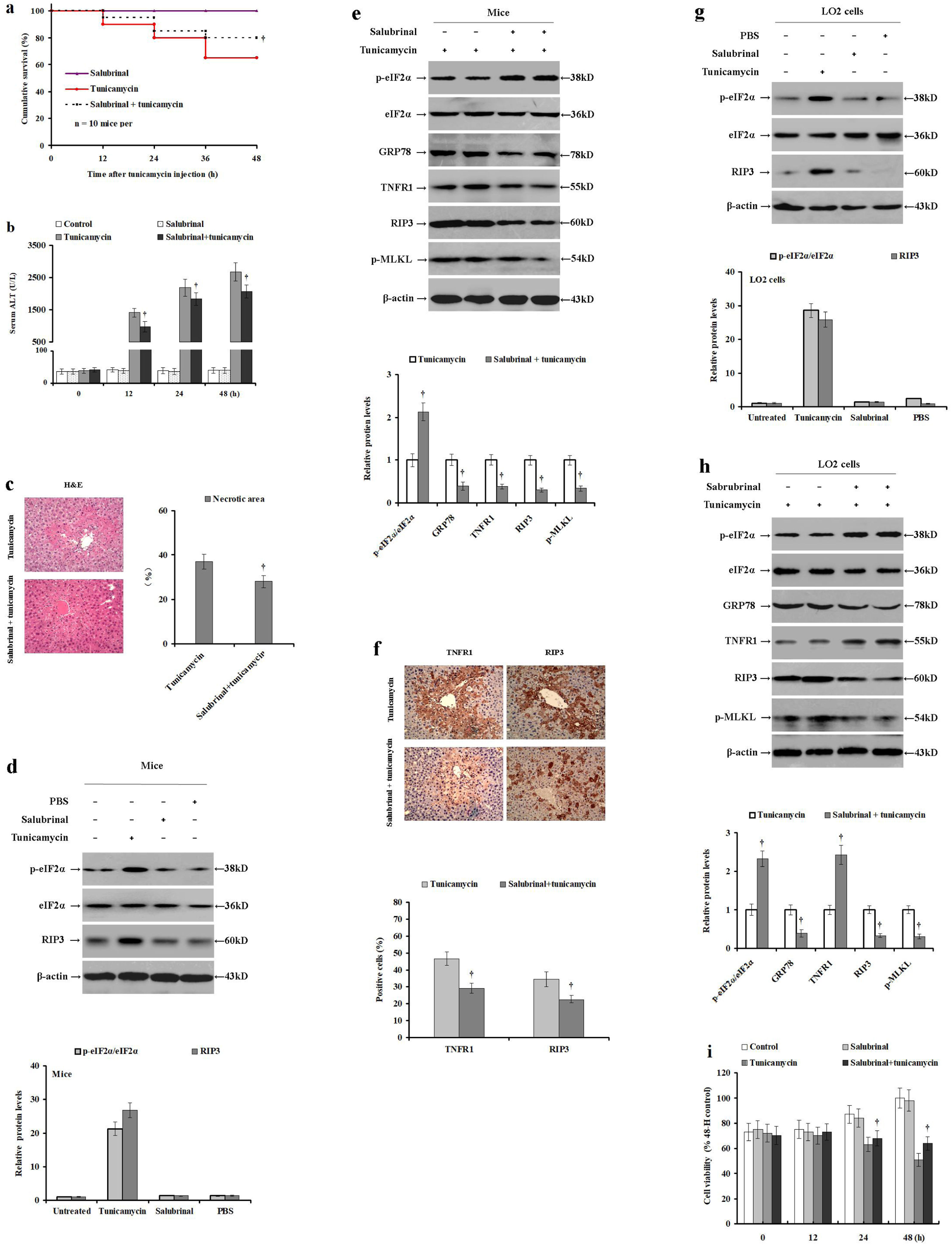
3.4Salubrinal pretreatment elevates tunicamycin-induced eIF2α phosphorylation and reduces ER stress and necroptosisIn cells, ER stress triggers eIF2α phosphorylation. Therefore, we used salubrinal to selectively inhibit eIF2α dephosphorylation and reduce ER stress to investigate the role of eIF2α phosphorylation in hepatocyte necroptosis and ER stress in an acute liver injury model. To this end, male BALB/c mice were pretreated with PBS or salubrinal before they were injected with tunicamycin or PBS. Compared to the tunicamycin treatment group, the cumulative survival was significantly improved in mice pretreated with salubrinal before tunicamycin injection (p<0.05; Fig. 4a). Tunicamycin treatment significantly increased serum ALT levels compared to the control or PBA treated, while tunicamycin with salubrinal pretreatment significantly reduced serum ALT levels (p<0.05; Fig. 4b). Salubrinal pretreatment before tunicamycin administration significantly reduced hepatocyte necrosis as verified by H&E staining (p<0.05; Fig. 4c). Importantly, salubrinal-alone did not change the cumulative mortality (Fig. 4a). Similarly, salubrinal treatment-alone did not change the serum ALT levels (Fig. 4b) or eIF2α phosphorylation and RIP3 expression (Fig. 4d) when compared to the control mice without liver injury. In contrast, salubrinal pretreatment before tunicamycin administration significantly increased intrahepatic eIF2α phosphorylation, and reduced TNFR1, RIP3, GRP78 expression, as well as MLKL phosphorylation (p<0.05; Fig. 4e), when compared to the tunicamycin-treated mice. In agreement, compared with tunicamycin-treatment, the relative expression levels of TNFR1 and RIP3 were reduced in the tunicamycin/salubrinal pretreatment group as verified by immunohistochemical staining (34.37±3.39% vs 22.53±2.18%; p<0.05; Fig. 4f). These data indicate that enhanced eIF2α phosphorylation mitigates tunicamycin-induced hepatocyte ER stress, TNFR1 expression, and necroptosis in mice.
 The cumulative survival of each mice group was investigated at 0, 12, 24, 36 and 48h (n=10 mice per group). (b) Serum ALT levels were detected using the rate method in the control (PBS and PBS), salubrinal (salubrinal pretreatment and PBS injection), tunicamycin (PBS and tunicamycin injection) and the salubrinal+tunicamycin (salubrinal pretreatment and tunicamycin injection) groups. (c) Histological examination of hepatocyte necrosis in the livers of tunicamycin and tunicamycin/salubrinal pretreated group (n=10 mice per group). (d) The relative expression of intrahepatic p-eIF2α, eIF2α and RIP3 in the untreated, tunicamycin (PBS and tunicamycin injection), control (PBS and PBS injection), and salubrinal (salubrinal pretreatment and PBS injection) mice. (e) The relative expression of intrahepatic TNFR1, phosphorylated eIF2α, total eIF2α, GRP78, RIP3, and phosphorylated MLKL in the salubrinal (salubrinal pretreatment and PBS injection) and the salubrinal+tunicamycin (salubrinal pretreatment and tunicamycin injection) mice. Protein samples were electrophoresed in duplicate lanes. (f) Immunohistochemical analysis of intrahepatic TNFR1 and RIP3 expression in the livers of salubrinal (salubrinal pretreatment and PBS injection) and the salubrinal+tunicamycin (salubrinal pretreatment and tunicamycin injection) groups (magnification 100×). (g) The relative expression of p-eIF2α, eIF2α and RIP3 in the control (PBS and PBS incubation), tunicamycin (PBS and tunicamycin incubation) and salubrinal (salubrinal pretreatment and PBS incubation; duplicate lanes are presented) LO2 cells. (h) The relative levels of TNFR1, p-eIF2α, eIF2α, GRP78, RIP3, and p-MLKL expression in the tunicamycin (PBS and tunicamycin incubation) and salubrinal/tunicamycin (salubrinal pretreatment and tunicamycin incubation) LO2 cells. Protein samples were electrophoresed in duplicate lanes. (i) Viability of LO2 cells determined by MTS assay in the control (PBS and PBS incubation), salubrinal (salubrinal pretreatment and PBS incubation), tunicamycin (PBS and tunicamycin incubation) and the salubrinal+tunicamycin (salubrinal pretreatment and tunicamycin incubation) groups. Histograms represent mean±SD of four independent experiments. †p<0.05 versus the tunicamycin group.")
Salubrinal pretreatment elevates tunicamycin-induced eIF2α phosphorylation and mitigates hepatocyte ER stress and necroptosis. Male BALB/c mice were pretreated with PBS or salubrinal for 2h, and then injected with PBS or tunicamycin for 24h. LO2 cells were pretreated with PBS or salubrinal for 2h, and then incubated with PBS or tunicamycin. (a) The cumulative survival of each mice group was investigated at 0, 12, 24, 36 and 48h (n=10 mice per group). (b) Serum ALT levels were detected using the rate method in the control (PBS and PBS), salubrinal (salubrinal pretreatment and PBS injection), tunicamycin (PBS and tunicamycin injection) and the salubrinal+tunicamycin (salubrinal pretreatment and tunicamycin injection) groups. (c) Histological examination of hepatocyte necrosis in the livers of tunicamycin and tunicamycin/salubrinal pretreated group (n=10 mice per group). (d) The relative expression of intrahepatic p-eIF2α, eIF2α and RIP3 in the untreated, tunicamycin (PBS and tunicamycin injection), control (PBS and PBS injection), and salubrinal (salubrinal pretreatment and PBS injection) mice. (e) The relative expression of intrahepatic TNFR1, phosphorylated eIF2α, total eIF2α, GRP78, RIP3, and phosphorylated MLKL in the salubrinal (salubrinal pretreatment and PBS injection) and the salubrinal+tunicamycin (salubrinal pretreatment and tunicamycin injection) mice. Protein samples were electrophoresed in duplicate lanes. (f) Immunohistochemical analysis of intrahepatic TNFR1 and RIP3 expression in the livers of salubrinal (salubrinal pretreatment and PBS injection) and the salubrinal+tunicamycin (salubrinal pretreatment and tunicamycin injection) groups (magnification 100×). (g) The relative expression of p-eIF2α, eIF2α and RIP3 in the control (PBS and PBS incubation), tunicamycin (PBS and tunicamycin incubation) and salubrinal (salubrinal pretreatment and PBS incubation; duplicate lanes are presented) LO2 cells. (h) The relative levels of TNFR1, p-eIF2α, eIF2α, GRP78, RIP3, and p-MLKL expression in the tunicamycin (PBS and tunicamycin incubation) and salubrinal/tunicamycin (salubrinal pretreatment and tunicamycin incubation) LO2 cells. Protein samples were electrophoresed in duplicate lanes. (i) Viability of LO2 cells determined by MTS assay in the control (PBS and PBS incubation), salubrinal (salubrinal pretreatment and PBS incubation), tunicamycin (PBS and tunicamycin incubation) and the salubrinal+tunicamycin (salubrinal pretreatment and tunicamycin incubation) groups. Histograms represent mean±SD of four independent experiments. †p<0.05 versus the tunicamycin group.
Next, we repeated the same model in LO2 cells to further investigate the impact of eIF2α phosphorylation on ER stress and necroptosis in vitro. LO2 cells were pretreated with PBS or salubrinal, and then incubated with tunicamycin or PBS. Salubrinal treatment-alone did not change the eIF2α phosphorylation or RIP3 expression in LO2 cells without ER stress (Fig. 4g). However, salubrinal pretreatment before tunicamycin significantly increased eIF2α phosphorylation and reduced the CHOP and RIP3 expression (p<0.05), as well as MLKL phosphorylation levels, as compared to tunicamycin-treated cells (p<0.05; Fig. 4h). In addition, it partially restored TNFR1 expression and LO2 cell viability (p<0.05; Fig. 4i). These data confirm that eIF2α phosphorylation mitigated tunicamycin-induced ER stress and necroptosis in LO2 cells.
4DiscussionIn the present study, we demonstrated that tunicamycin and d-galactosamine successfully induced ER stress and hepatocyte necroptosis, as well as eIF2α phosphorylation, in BALB/c mice and LO2 cells. Moreover, d-galactosamine could induce ER stress and necroptosis without up-regulating TNFR1 expression, indicating that ER stress and necroptosis can be mediated via TNFα/TNFR1-independent pathways in hepatocytes. Pretreatment with PBA mitigated tunicamycin-induced hepatocyte necroptosis in mice and LO2 cells by alleviating ER stress, while salubrinal pretreatment mitigated tunicamycin-induced ER stress and hepatocyte necroptosis by inhibiting eIF2α dephosphorylation and increasing p-eIF2α levels. In addition, PBA and salubrinal pretreatment alleviated ER stress and necroptosis, but it partially restored TNFR1 expression in LO2 cells challenged with tunicamycin. Together, these results demonstrate the crucial role of eIF2α phosphorylation in mitigating ER stress and necroptosis independent of TNFR1 signaling in an acute liver injury model. To the best of our knowledge, this is the first study that reveals the beneficial role of eIF2α phosphorylation in alleviating hepatocyte necroptosis and ER stress in an acute liver injury.
TNF-α is a pleiotropic cytokine involved in inflammation and cell injury [39]. TNF-α function is mediated by TNFR1 signaling [30,40]. TNFR1 mediates cell injury via the “death domain” and up-regulates the TNFR1 expression may enhance the cell sensitivity to TNF-α-induced cell injury. Effective control of ER stress can mitigate liver injury [41]. Our results demonstrate that ER stress inhibits TNFR1 expression in tunicamycin-incubated LO2 cells and d-galactosamine-treated mice, suggesting that TNFR1 signaling may not be necessary to trigger hepatocyte necrosis. Moreover, tunicamycin or d-galactosamine administration significantly induced hepatocyte ER stress and necroptosis in mice. PBA pretreatment significantly mitigated the tunicamycin-induced hepatocyte ER stress and necroptosis in mice. In addition, tunicamycin treatment induced ER stress and necroptosis, and reduced LO2 cells viability. These effects were moderated by PBA pretreatment. These results not only support that ER stress mediates hepatocyte necroptosis, but also suggest that ER stress can induce necroptosis independent of TNFR1 signaling in acute liver injury [15]. Moreover, our data showed that ER stress affects TNFR1 expression differently, indicating that ER stress may have multiple ways to regulate TNFR1 expression. Future investigations should focus on dissecting the underlying molecular mechanism.
eIF2α phosphorylation can inhibit protein synthesis and up-regulate ATF4 expression [42]. Inhibition of protein synthesis can reduce the protein folding load in the ER [43]. In this study, tunicamycin and d-galactosamine significantly induced hepatocyte ER stress and eIF2α phosphorylation. Interestingly, pretreatment with salubrinal significantly elevated eIF2α phosphorylation and reduced tunicamycin-induced ER stress, as well as necroptosis, in mice and LO2 cells. On the other hand, PBA pretreatment decreased eIF2α phosphorylation and necroptosis. These data suggest that eIF2α phosphorylation does not directly modulate hepatocyte necroptosis, but it indirectly mitigates tunicamycin-induced ER stress and regulates hepatocyte necroptosis. This discrepancy can be attributed to the fact that PBA acts as a chemical molecular chaperone that reduces ER stress by reducing the need for eIF2α phosphorylation [44]. Previously, Wang et al. demonstrated that up-regulating phosphorylated eIF2α by salubrinal administration attenuated ER stress and improved necroptosis [45].
Hepatocyte necroptosis has been implicated in different acute and chronic pathological conditions, including NAFLD or NASH, alcohol-induced liver injury, drug-induced liver injury, as well as hepatitis B and C viral infections [46]. ER stress also has been implicated in a variety of liver diseases, including NAFLD, alcohol-induced liver injury, drug-induced liver injury, hepatic insulin resistance, ischemia-reperfusion injury and hepatitis viral infections [47–49]. However, whether ER stress is directly regulated to necroptosis has not been fully elucidated. Our data shows that inhibition of ER stress can reduce hepatocyte necroptosis by up-regulating eIF2α phosphorylation, thus decreasing liver injury. This mechanism may also explain the relationship of ER stress and necroptosis in other liver diseases besides liver injury, which may be a therapeutic target for the treatment of liver diseases.
Taken together, we observed that the selective elevation of eIF2α phosphorylation could regulate ER stress through a feedback mechanism, which consequently reduces hepatocyte necroptosis, thus mitigating the acute liver injury. These results provide new insights into the underlying molecular mechanisms that regulate acute liver injury.
List of abbreviationsATF4 activating transcription factor 4 activating transcription factor 6 alanine aminotransferase C/EBP homologous protein endoplasmic reticulum eukaryotic initiation factor 2 alpha growth arrest and DNA damage 34 glucose-regulated protein 78 horseradish peroxidase hematoxylin and eosin inositol-requiring enzyme 1 alpha mixed lineage kinase domain-like pseudokinase protein phosphatase 1 polyvinylidene fluoride protein kinase R-like ER kinase phosphate buffer saline 4-phenylbutyric acid receptor-interacting protein 3
This study was partially supported by the National Natural Science Foundation of China (81560110), Tian Qing Liver Disease Research Fund Project of Chinese Foundation for Hepatitis Prevention and Control (TQGB20170050), as well as The Science and Technology Planning Projects of Guizhou Province (QKH·LH [2017] 7093, QKH·ZC[2019] 2803).
Conflict of interestThe authors declare that they have no conflicts of interest.



