






La hiperlipemia familiar combinada (HFC) es la hiperlipemia genética más común, y una causa frecuente de enfermedad cardiovascular prematura. Sin embargo, el origen genético de esta enfermedad todavía no se conoce totalmente. En el presente estudio, analizamos el perfil de expresión génica en monocitos de sangre periférica en un grupo de individuos control y en un grupo de pacientes con HFC antes y después del tratamiento con atorvastatina (ATV), con el objetivo de estudiar y profundizar en los mecanismos genéticos asociados a esta enfermedad, e identificar posibles dianas terapéuticas nuevas.
Métodos y resultadosSe aislaron monocitos de sangre periférica de individuos control y de pacientes con HFC, del mismo sexo y edad, antes y después del tratamiento con ATV. El perfil de expresión génica se obtuvo usando los microarrays GeneChip Human Genome U133A de Affymetrix, y los cambios en la expresión de los genes seleccionados se validaron posteriormente con la técnica de retro-transcripción de la reacción en cadena de la polimerasa a tiempo real. Nuestros resultados mostraron cambios en la expresión de numerosos genes implicados en funciones clave del macrófago, especialmente en la respuesta inflamatoria (MNDA, IL1R2, ALCAM, LRIG1 y DR3), el control de la composición de la matriz extracelular (FN1, SDC2 y TFPI2) y el metabolismo lipídico (CD36).
ConclusionesNuestros resultados indican que la HFC afecta de forma directa o indirecta al perfil de expresión génica en monocitos de sangre periférica. Los cambios en la expresión de algunos genes (MNDA, CD36, TFPI2, LRIG1 y DR3) podrían atribuirse al entorno proinflamatorio que rodea los monocitos en la HFC, que es parcialmente revertido por el tratamiento con ATV. Por otra parte, los cambios en la expresión génica que no revierten tras el tratamiento con ATV podrían relacionarse con anomalías metabólicas del tejido adiposo (valores bajos de adiponectina y elevación de ácidos grasos libres y triglicéridos) que no se corrigen con este tratamiento.
The genetic origin of familial combined hyperlipidemia (FCH) is not well understood. We used peripheral blood monocytes as a suitable target for differential gene expression assessment in FCH.
Methods and resultsPeripheral blood monocytes were isolated from male FCH patients basally and after atorvastatin treatment. Sex-, age- and adiposity-matched controls were also studied. Analysis of gene expression was performed using the GeneChip Human Genome U133A microarrays. Changes in the expression of selected genes were confirmed by real time RT-PCR. Our results showed the differential expression of genes involved in key macrophage functions, specially regarding the inflammatory response (MNDA, IL1R2, ALCAM, LRIG1 and DR3), extracellular matrix control (FN1, SDC2 and TFPI2) and lipid metabolism (CD36).
Conclusions.Monocyte gene expression profiling can provide insight into the pathogenesis of FCH. The results suggest that alterations in the expression of some genes (MNDA, CD36, TFPI2, LRIG1 and DR3) may be related to a proinflammatory environment in FCH monocytes, which is partially reversed by atorvastatin. However, metabolic dysfunction in adipose tissue may account for lower adiponectin and higher FFA and triglyceride plasma levels in FCH, which are not corrected by treatment. These abnormalities could also trigger changes in gene expression that atorvastatin cannot modify.
La hiperlipemia familiar combinada (HFC) es la hiperlipemia genética más común en humanos, ya que afecta al 1-3% de la población general, y se asocia a un riesgo muy elevado de presentar enfermedad cardiovascular prematura1 . El perfil lipídico de la HFC se caracteriza por hipercolesterolemia y/o hipertrigliceridemia, valores elevados de apolipoproteína (apo) B y de colesterol unido a lipoproteínas de baja densidad (cLDL), valores reducidos de colesterol unido a lipoproteínas de alta densidad (cHDL) y la presencia de lipoproteínas de baja densidad (LDL) pequeñas y densas (sdLDL)2. Básicamente, la anomalía metabólica presente en esta enfermedad es la sobreproducción de lipoproteínas de muy baja densidad (VLDL), que puede ser consecuencia de un incorrecto metabolismo de los ácidos grasos libres (AGL)3. El incremento del estrés oxidativo y la inflamación son otras características que se asocian con este trastorno que pueden contribuir a un desarrollo acelerado de aterosclerosis4.
Se han relacionado un cierto número de regiones cromosómicas (1q21-23, 11p14.1-q12.1 y 16q22-24.1) con la patogenia de la HFC`5, pero la base genética de esta enfermedad no se ha elucidado totalmente en la actualidad. Los arrays de expresión génica son herramientas muy útiles para estudiar los mecanismos moleculares subyacentes en la HFC, así como para aumentar el conocimiento sobre los mecanismos de acción de los fármacos utilizados actualmente para el tratamiento de esta enfermedad. Entre estos fármacos destacan los inhibidores de la 3-hidroxi-3-metilglutaril CoA reductasa, o estatinas. En el presente trabajo, se han estudiado las diferencias en el perfil de expresión génica en monocitos aislados de sangre periférica, procedentes de pacientes con HFC y de individuos sanos. Por otro lado, los pacientes con HFC se trataron con atorvastatina durante 4 semanas, período tras el cual se analizó el perfil de expresión génica nuevamente. El estudio se ha llevado a cabo en monocitos sanguíneos debido a que son células clave en el proceso aterogénico6.
MétodosPacientesSe seleccionó a 12 varones asintomáticos con HFC en la Unidad de Lípidos del Hospital Clínic de Barcelona. El diagnóstico de HFC se realizó según Eurlings et al7 en individuos con dislipemia mixta asociada a unos valores séricos de apo B 1,20 g/l; transmisión monogénica de hiperlipemia con fenotipo variable relativo a las lipoproteínas; ausencia de niños hipercolesterolémicos en la familia; historia familiar de enfermedad cardiovascular prematura, y ausencia del genotipo apo E 2/2. Se seleccionó a un grupo control de 12 varones sanos, con una edad y adiposidad similares al grupo HFC, entre los miembros del personal y los médicos del hospital. Todos los participantes dieron su consentimiento, previa información escrita del protocolo aprobado por el comité examinador local.
Diseño del estudioLos pacientes con HFC mantuvieron un período de 4 semanas previas al estudio en las cuales no tomaron ningún fármaco hipolipemiante. Pasado este período, se trataron con atorvastatina (40 mg/día) durante 4 semanas. Las muestras sanguíneas se recogieron una única vez en el grupo control, mientras que en los pacientes HFC se recogieron muestras de sangre al principio y después de las 4 semanas de tratamiento con atorvastatina.
Parámetros plasmáticosA excepción de la determinación de glucosa sanguínea y de lípidos, que se realizó en el momento de la extracción, las muestras séricas y plasmáticas se conservaron a −80 °C, utilizando ácido edético (EDTA, del inglés ethylene diamine tetraacetic) como anticoagulante, para analizarlas al final del estudio. La glucosa sérica se analizó con el método de la glucosa oxidasa. El colesterol y los triglicéridos se cuantificaron utilizando métodos enzimáticos. El cHDL se determinó por precipitación con ácido fosfotúngstico y cloruro de magnesio. El cLDL se estimó utilizando la ecuación de Friedewald et al8, excepto en los pacientes con triglicéridos > 300 mg/dl, en los que éste se midió mediante ultracentrifugación7. Las apo A-1 y B y la lipoproteína A se determinaron por turbidimetría. En los individuos con diagnóstico clínico de HFC, el genotipo de apo E se determinó por el método de Wenham et al9. Los AGL se determinaron por métodos colorimétricos enzimáticos (Wako, Neuss, Alemania). La leptina y la adiponectina se determinaron por ensayo radioinmunológico (Linco Research, St Charles, MO, Estados Unidos), y la proteína C reactiva mediante inmunofelometría. La fibronectina (FN) y los receptores solubles de factor de necrosis tumoral (sTNF-R) se cuantificaron por técnicas de ELISA (Bender MedSystems GmbH, Viena, Austria). Todos los análisis se realizaron por duplicado.
Obtención de monocitos a partir de sangre periféricaLa fracción celular mononucleada se aisló a partir de 50 ml de sangre periférica mediante centrifugación en gradiente de densidad mediante el reactivo Ficoll-Paque (Amersham Biosciences Corp., Pisacataway, Estados Unidos). Las células se lavaron con tampón fosfato salino (PBS) sin calcio ni magnesio, pH 7,4, y se resuspendieron en medio RPMI-1640 (25 mM HEPES, 100 U/ml de penicilina, 100 mg/ml de estreptomicina, 1% de glutamina, 1% de aminoácidos no esenciales y 2% de piruvato sódico). Para purificar los monocitos por adhesión en placas de cultivo, las células se sembraron a una densidad de 0,8 × 106 células/ml, y después de 2 h a 37 °C a un 5% de CO2, se lavaron con PBS con el fin de eliminar las células no adherentes. Este método proporcionó unas preparaciones de monocitos con una pureza superior al 92%, como se pudo comprobar por citometría de flujo usando un sistema bicolor (CD14-FITC/CD64-PE) de Santa Cruz Biotechnology (Santa Cruz, Estados Unidos).
Aislamiento del ácido ribonucleico total e hibridación con el arrayEl ARN total se aisló de los monocitos usando Trizol (Invitrogen Corp.), y posteriormente se purificó usando el kit RNeasy (Qiagen Inc, Valencia, Estados Unidos), cuantificado por espectofotometría a 260 nm, y su pureza se comprobó a partir de la relación de absorbancias a 260–280 nm. La integridad del ARN se comprobó mediante electroforesis en gel de agarosa.
Se prepararon un total de 9 grupos de muestras (pools), 3 por cada condición: control (CT), HFC antes del tratamiento (HFC) y después del tratamiento con atorvastatina (HFC + ATV). Cada pool se preparó con la misma cantidad de ARN procedente de cultivos individuales de monocitos. Los pools de ARN se utilizaron para analizar el perfil de expresión génica usando el Gene-Chip Human Genome U133A 2.0 Array de Affymetrix.
Procesamiento de los datos y análisis estadísticoLos arrays se escanearon y las imágenes obtenidas se cuantificaron de acuerdo con el procedimiento estándar de Affymetrix, con el fin de obtener la intensidad de señal para cada gen en cada array. Los valores de expresión se normalizaron utilizando el método RMA (del inglés robust multi-array average)10. Antes de analizar los datos, éstos se filtraron para eliminar genes de señal baja y genes de variabilidad baja. La selección de genes diferencialmente expresados en las diferentes condiciones se basó en un modelo lineal de análisis con moderación empírica de Bayes de las variancias estimadas, siguiendo la metodología desarrollada por Smyth11. Se consideraron diferencialmente expresados los genes cuya variación mostró un valor de p ≤ 0,01.
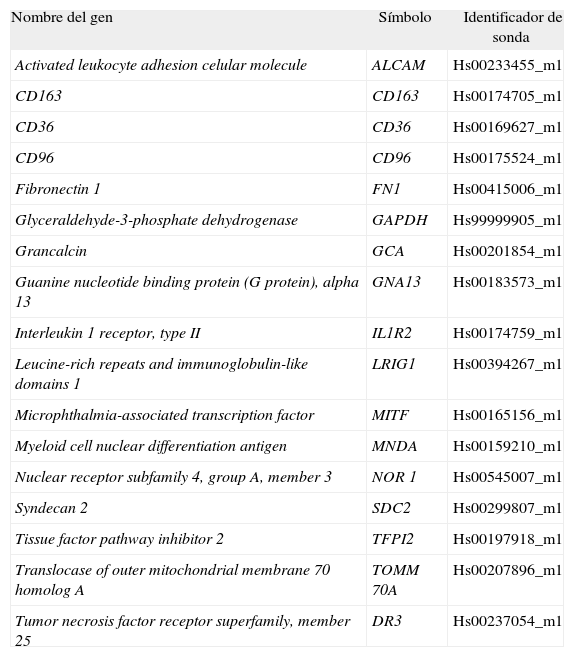
Retro-transcripción de la reacción en cadena de la polimerasa a tiempo realCon el fin de confirmar los patrones de expresión de los genes sobreexpresados y reprimidos, se seleccionaron una serie de genes para un análisis posterior usando la técnica de retro-transcripción de la reacción en cadena de la polimerasa (PCR) a tiempo real. Para ello, el ácido desoxirribonucleico (ADN) complementario se sintetizó a partir de las mismas muestras de ARN utilizadas en los arrays, mezclando 0,5 μg de ARN total, 125 ng de random hexamers en presencia de 75 mM KCl, 3 mM MgCl2, 10 mM dithiothreitol, 200 U de transcriptasa reversa (Moloney murine leukemia virus reverse transcriptase), 20 U de ARNsin, 0,5 mM de cada dNTP (Sigma), y tampón Tris–HCl 50 mM (pH 8,3). Las muestras se incubaron a 37 °C durante 60 min. La PCR a tiempo real se realizó utilizando un sistema Perkin-Elmer ABI Prism 7700 y sondas TaqMan (tabla 1) procedentes de Applied Biosystems, Foster City, Estados Unidos. Como control interno, se utilizó el gen de la gliceraldehído-3-fosfato deshidrogenasa. Para el análisis de los datos se utilizó el sequence detector software (SDS 1.9.1). Para cada curva de amplificación se obtuvo un valor de umbral (CT), y después de normalizar por el gen de referencia GAPDH, se calculó la relación de expresiones para cada gen, basada en la diferencia entre las CT de la muestra y el control correspondiente. El análisis estadístico se realizó por el método t test utilizando el programa GraphPad Instat.
Sondas TaqMan®
| Nombre del gen | Símbolo | Identificador de sonda |
| Activated leukocyte adhesion celular molecule | ALCAM | Hs00233455_m1 |
| CD163 | CD163 | Hs00174705_m1 |
| CD36 | CD36 | Hs00169627_m1 |
| CD96 | CD96 | Hs00175524_m1 |
| Fibronectin 1 | FN1 | Hs00415006_m1 |
| Glyceraldehyde-3-phosphate dehydrogenase | GAPDH | Hs99999905_m1 |
| Grancalcin | GCA | Hs00201854_m1 |
| Guanine nucleotide binding protein (G protein), alpha 13 | GNA13 | Hs00183573_m1 |
| Interleukin 1 receptor, type II | IL1R2 | Hs00174759_m1 |
| Leucine-rich repeats and immunoglobulin-like domains 1 | LRIG1 | Hs00394267_m1 |
| Microphthalmia-associated transcription factor | MITF | Hs00165156_m1 |
| Myeloid cell nuclear differentiation antigen | MNDA | Hs00159210_m1 |
| Nuclear receptor subfamily 4, group A, member 3 | NOR 1 | Hs00545007_m1 |
| Syndecan 2 | SDC2 | Hs00299807_m1 |
| Tissue factor pathway inhibitor 2 | TFPI2 | Hs00197918_m1 |
| Translocase of outer mitochondrial membrane 70 homolog A | TOMM 70A | Hs00207896_m1 |
| Tumor necrosis factor receptor superfamily, member 25 | DR3 | Hs00237054_m1 |
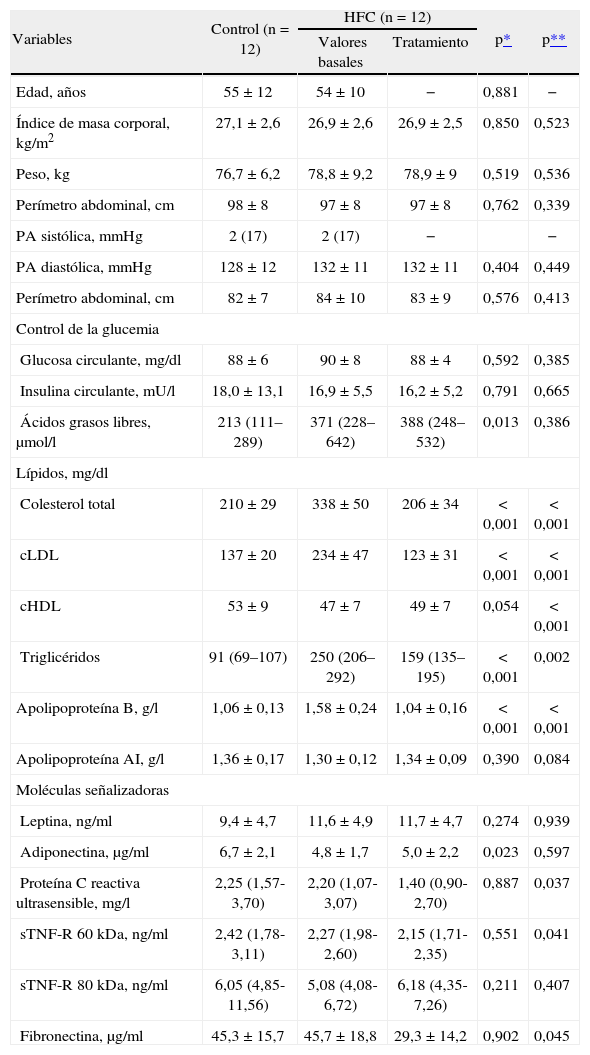
En la tabla 2 se resumen las características clínicas de los 2 grupos del estudio. Los pacientes HFC y los individuos control presentaron valores similares de presión sanguínea y de glucemia, pero diferencias marcadas en cuanto al perfil lipídico. Los valores de AGL fueron más altos en el grupo HFC que en los CT, mientras que las concentraciones plasmáticas de adiponectina resultaron ser menores.
Características de los grupos del estudio y efectos del tratamiento durante 4 semanas con atorvastatina en los pacientes de hiperlipemia familiar combinada
| Variables | Control (n = 12) | HFC (n = 12) | p* | p** | |
| Valores basales | Tratamiento | ||||
| Edad, años | 55 ± 12 | 54 ± 10 | − | 0,881 | − |
| Índice de masa corporal, kg/m2 | 27,1 ±2,6 | 26,9 ± 2,6 | 26,9 ± 2,5 | 0,850 | 0,523 |
| Peso, kg | 76,7 ± 6,2 | 78,8 ± 9,2 | 78,9 ± 9 | 0,519 | 0,536 |
| Perímetro abdominal, cm | 98 ± 8 | 97 ± 8 | 97 ± 8 | 0,762 | 0,339 |
| PA sistólica, mmHg | 2 (17) | 2 (17) | − | − | |
| PA diastólica, mmHg | 128 ± 12 | 132 ± 11 | 132 ± 11 | 0,404 | 0,449 |
| Perímetro abdominal, cm | 82 ± 7 | 84 ± 10 | 83 ± 9 | 0,576 | 0,413 |
| Control de la glucemia | |||||
| Glucosa circulante, mg/dl | 88 ± 6 | 90 ± 8 | 88 ± 4 | 0,592 | 0,385 |
| Insulina circulante, mU/l | 18,0 ± 13,1 | 16,9 ±5,5 | 16,2 ±5,2 | 0,791 | 0,665 |
| Ácidos grasos libres, μmol/l | 213 (111–289) | 371 (228–642) | 388 (248–532) | 0,013 | 0,386 |
| Lípidos, mg/dl | |||||
| Colesterol total | 210 ± 29 | 338 ± 50 | 206 ± 34 | < 0,001 | < 0,001 |
| cLDL | 137 ± 20 | 234 ± 47 | 123 ±31 | < 0,001 | < 0,001 |
| cHDL | 53 ± 9 | 47 ± 7 | 49 ± 7 | 0,054 | < 0,001 |
| Triglicéridos | 91 (69–107) | 250 (206–292) | 159 (135–195) | < 0,001 | 0,002 |
| Apolipoproteína B, g/l | 1,06 ±0,13 | 1,58 ±0,24 | 1,04 ±0,16 | < 0,001 | < 0,001 |
| Apolipoproteína AI, g/l | 1,36 ±0,17 | 1,30 ±0,12 | 1,34 ± 0,09 | 0,390 | 0,084 |
| Moléculas señalizadoras | |||||
| Leptina, ng/ml | 9,4 ± 4,7 | 11,6 ±4,9 | 11,7 ±4,7 | 0,274 | 0,939 |
| Adiponectina, μg/ml | 6,7 ±2,1 | 4,8 ± 1,7 | 5,0 ± 2,2 | 0,023 | 0,597 |
| Proteína C reactiva ultrasensible, mg/l | 2,25 (1,57-3,70) | 2,20 (1,07-3,07) | 1,40 (0,90-2,70) | 0,887 | 0,037 |
| sTNF-R 60 kDa, ng/ml | 2,42 (1,78-3,11) | 2,27 (1,98-2,60) | 2,15 (1,71-2,35) | 0,551 | 0,041 |
| sTNF-R 80 kDa, ng/ml | 6,05 (4,85-11,56) | 5,08 (4,08-6,72) | 6,18 (4,35-7,26) | 0,211 | 0,407 |
| Fibronectina, μg/ml | 45,3 ± 15,7 | 45,7 ± 18,8 | 29,3 ± 14,2 | 0,902 | 0,045 |
cHDL: colesterol unido a lipoproteínas de alta densidad; cLDL: colesterol unido a lipoproteínas de baja densidad; HFC: hiperlipemia familiar combinada; PA: presión arterial; sTNF-R: receptor soluble del factor de necrosis tumoral.
Datos expresados como media ± desviación estándar o mediana (rango intercuartílico).
En la tabla 2 se muestran los cambios en las concentraciones de lípidos y de lipoproteínas séricas en el grupo HFC después de 4 semanas de tratamiento con atorvastatina a una dosis de 40 mg/día. Los cambios en el colesterol total (−39%), cLDL (−47%), cHDL (4%), triglicéridos (−36%), y apo B (−34%) fueron todos estadísticamente significativos. No se observaron cambios por lo que respecta a adiposidad, presión sanguínea, glucemia y valores circulantes de leptina y adiponectina.
Genes diferencialmente expresadosNuestros resultados mostraron la expresión diferencial de 82 genes diferencialmente expresados en monocitos de pacientes HFC respecto a los del grupo CT, de los cuales 46 se encontraban reprimidos y 36, sobreexpresados. Cuando se comparó el perfil de expresión génica en monocitos de pacientes HFC antes y transcurridas las 4 semanas de tratamiento con atorvastatina (HFC + ATV frente a HFC), se hallaron 88 genes diferencialmente expresados, 36 reprimidos y 52 sobreexpresados.
Con el objetivo de validar estos resultados, se seleccionaron diversos genes, a partir del cambio de expresión observado y/o a su función, para un análisis posterior mediante PCR a tiempo real (tablas 3 y 4).
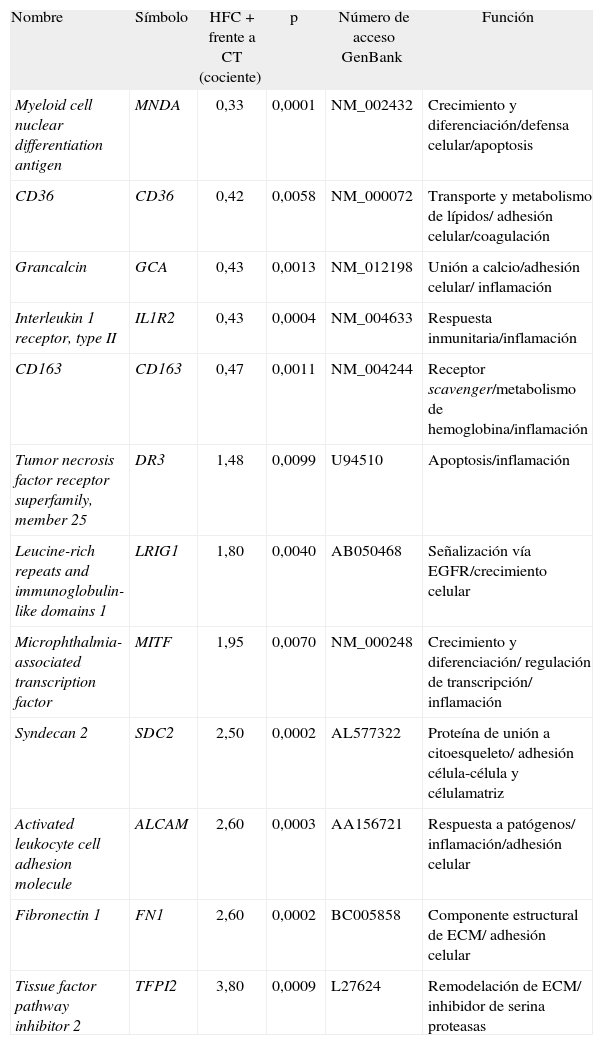
Selección de genes diferencialmente expresados en monocitos de pacientes con hiperlipemia familiar combinada respecto a los individuos control
| Nombre | Símbolo | HFC + frente a CT (cociente) | p | Número de acceso GenBank | Función |
| Myeloid cell nuclear differentiation antigen | MNDA | 0,33 | 0,0001 | NM_002432 | Crecimiento y diferenciación/defensa celular/apoptosis |
| CD36 | CD36 | 0,42 | 0,0058 | NM_000072 | Transporte y metabolismo de lípidos/ adhesión celular/coagulación |
| Grancalcin | GCA | 0,43 | 0,0013 | NM_012198 | Unión a calcio/adhesión celular/ inflamación |
| Interleukin 1 receptor, type II | IL1R2 | 0,43 | 0,0004 | NM_004633 | Respuesta inmunitaria/inflamación |
| CD163 | CD163 | 0,47 | 0,0011 | NM_004244 | Receptor scavenger/metabolismo de hemoglobina/inflamación |
| Tumor necrosis factor receptor superfamily, member 25 | DR3 | 1,48 | 0,0099 | U94510 | Apoptosis/inflamación |
| Leucine-rich repeats and immunoglobulin-like domains 1 | LRIG1 | 1,80 | 0,0040 | AB050468 | Señalización vía EGFR/crecimiento celular |
| Microphthalmia-associated transcription factor | MITF | 1,95 | 0,0070 | NM_000248 | Crecimiento y diferenciación/ regulación de transcripción/ inflamación |
| Syndecan 2 | SDC2 | 2,50 | 0,0002 | AL577322 | Proteína de unión a citoesqueleto/ adhesión célula-célula y célulamatriz |
| Activated leukocyte cell adhesion molecule | ALCAM | 2,60 | 0,0003 | AA156721 | Respuesta a patógenos/ inflamación/adhesión celular |
| Fibronectin 1 | FN1 | 2,60 | 0,0002 | BC005858 | Componente estructural de ECM/ adhesión celular |
| Tissue factor pathway inhibitor 2 | TFPI2 | 3,80 | 0,0009 | L27624 | Remodelación de ECM/ inhibidor de serina proteasas |
La tabla muestra el nombre, la expresión relativa respecto la situación control, el valor de p y el número de acceso de GenBank de una selección de genes que resultaron expresados diferencialmente en monocitos de pacientes con HFC respecto a los controles. La última columna indica la categoría funcional a la que pertenece cada gen. Los resultados son la media de 3 experimentos independientes realizados para cada condición.
CT: individuos control; ECM: extracellular matrix; EGFR: receptor para el factor de crecimiento epidérmico; HFC: hiperlipemia familiar combinada.
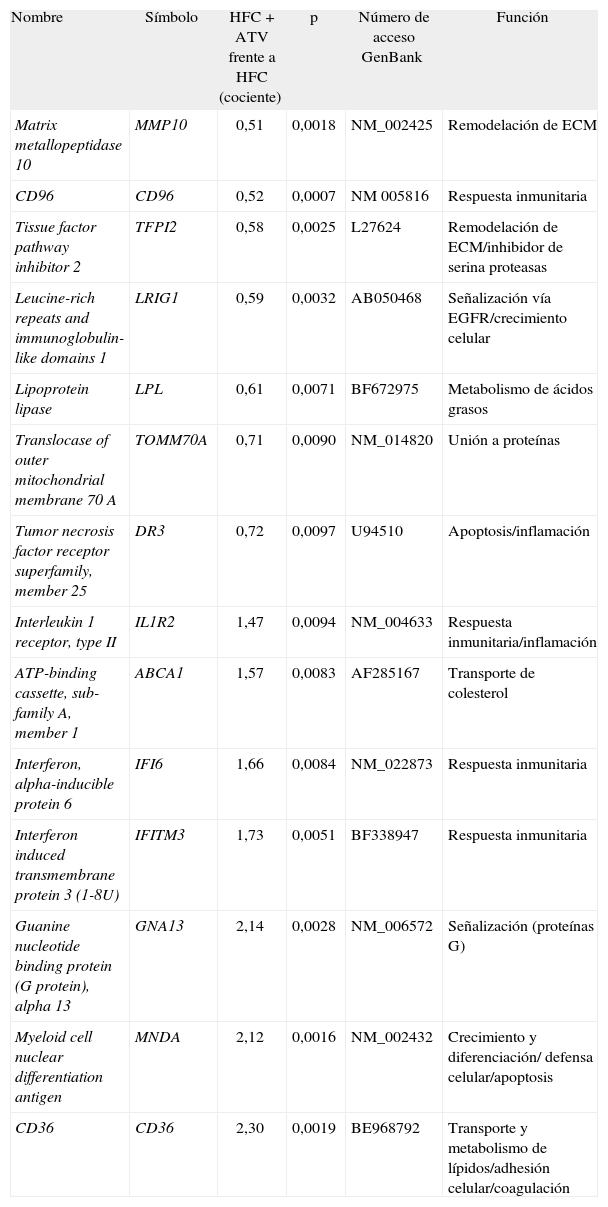
Selección de genes diferencialmente expresados en monocitos de pacientes con hiperlipemia familiar combinada (HFC) después del tratamiento con atorvastatina (HFC + ATV) comparados con la situación basal (HFC)
| Nombre | Símbolo | HFC + ATV frente a HFC (cociente) | p | Número de acceso GenBank | Función |
| Matrix metallopeptidase 10 | MMP10 | 0,51 | 0,0018 | NM_002425 | Remodelación de ECM |
| CD96 | CD96 | 0,52 | 0,0007 | NM 005816 | Respuesta inmunitaria |
| Tissue factor pathway inhibitor 2 | TFPI2 | 0,58 | 0,0025 | L27624 | Remodelación de ECM/inhibidor de serina proteasas |
| Leucine-rich repeats and immunoglobulin-like domains 1 | LRIG1 | 0,59 | 0,0032 | AB050468 | Señalización vía EGFR/crecimiento celular |
| Lipoprotein lipase | LPL | 0,61 | 0,0071 | BF672975 | Metabolismo de ácidos grasos |
| Translocase of outer mitochondrial membrane 70 A | TOMM70A | 0,71 | 0,0090 | NM_014820 | Unión a proteínas |
| Tumor necrosis factor receptor superfamily, member 25 | DR3 | 0,72 | 0,0097 | U94510 | Apoptosis/inflamación |
| Interleukin 1 receptor, type II | IL1R2 | 1,47 | 0,0094 | NM_004633 | Respuesta inmunitaria/inflamación |
| ATP-binding cassette, sub-family A, member 1 | ABCA1 | 1,57 | 0,0083 | AF285167 | Transporte de colesterol |
| Interferon, alpha-inducible protein 6 | IFI6 | 1,66 | 0,0084 | NM_022873 | Respuesta inmunitaria |
| Interferon induced transmembrane protein 3 (1-8U) | IFITM3 | 1,73 | 0,0051 | BF338947 | Respuesta inmunitaria |
| Guanine nucleotide binding protein (G protein), alpha 13 | GNA13 | 2,14 | 0,0028 | NM_006572 | Señalización (proteínas G) |
| Myeloid cell nuclear differentiation antigen | MNDA | 2,12 | 0,0016 | NM_002432 | Crecimiento y diferenciación/ defensa celular/apoptosis |
| CD36 | CD36 | 2,30 | 0,0019 | BE968792 | Transporte y metabolismo de lípidos/adhesión celular/coagulación |
La tabla muestra el nombre, la expresión relativa respecto la situación control, el valor de p y el número de acceso de GenBank de una selección de genes que resultaron expresados diferencialmente en monocitos de pacientes con HFC respecto a los controles. La última columna indica la categoría funcional a la que pertenece cada gen. Los resultados son la media de 3 experimentos independientes realizados para cada condición.
ECM: extracellular matrix; EGRR: receptor para el factor de crecimiento epidérmico.
Uno de los genes más reprimidos en los pacientes HFC según el análisis de los arrays fue myeloid cell nuclear differentiation antigen (MNDA), con una relación de expresión génica entre HFC y CT (cociente) de 0,33. Los resultados obtenidos por PCR a tiempo real confirmaron la disminución de expresión de MNDA (cociente de 0,42). También se utilizó este método para confirmar la represión del receptor de tipo 2 de la interleucina 1 (IL1R2), con un cociente del array de 0,43, y un cociente de PCR de 0,37. Otros genes cuya expresión resultó disminuida fueron grancalcina (cociente del array 0,43, cociente de PCR 0,46), CD36 (cociente del array 0,42, cociente de PCR 0,52) y CD163 (cociente de 0,47, en ambos casos).
Diversos genes relacionados con la matriz extracelular, y el desarrollo y estabilidad de la placa de ateroma también resultaron modificados en los pacientes HFC respecto al grupo CT. Así, la expresión del gen inhibidor de la vía del factor tisular 2 (TFPI 2), resultó incrementada 3,8 veces según los resultados del array, y 6,5 veces según los resultados obtenidos por PCR a tiempo real. También resultaron sobreexpresados los genes FN 1 (cociente del array 2,6, cociente de PCR 2,45) y syndecan 2 (SDC2, cociente del array 2,5, cociente de PCR 2,49). Otros genes cuya expresión fue significativamente más alta en muestras de monocitos de pacientes con HFC que en controles fueron activated leukocyte cell adhesion molecule (ALCAM, cociente de array 2,6, cociente de PCR 2,76), leucine-rich repeats and immunoglobin-like domains (LRIG1, cociente del array 1,80, cociente de PCR 2,52), y microphtalmia-associated transcription factor (MITF, cociente del array 1,95, cociente de PCR 2,55). Además, se analizó la expresión del miembro 25 de la superfamilia de receptores de TNF (TNFRSF 25, también denominado death receptor 3 [DR3]), que resultó incrementada por ambos métodos (cociente del array 1,48, cociente de PCR 1,84).
Genes diferencialmente expresados en monocitos de pacientes HFC después del tratamiento con atorvastatinaTres de los genes que se sobreexpresaron en los pacientes con HFC, TFPI2, LRIG1 y DR3, resultaron reprimidos en respuesta al tratamiento con atorvastatina. Así, según los resultados obtenidos a partir del análisis de arrays, la relación de expresión entre los grupos HFC + ATV y HFC (cociente) fue de 0,58, 0,59 y 0,72, respectivamente. Estos resultados se confirmaron por PCR a tiempo real (cociente de 0,29 para TFPI2 y 0,51 para LRIG1 y DR3). Otros genes reprimidos debido al tratamiento y elegidos para la validación fueron CD96 y la translocasa de la membrana externa mitocondrial 70A (TOMM70A) (cociente de array 0,52 y 0,71, respectivamente). Para ambos genes, se observó también una menor expresión cuando se analizó por PCR, aunque de forma menos evidente (0,73 y 0,81, respectivamente).
Por otra parte, la expresión de los genes CD36 y MNDA, que es más baja en muestras de HFC respecto los CT, resultó incrementada tras el tratamiento con atorvastatina. Estos resultados también se confirmaron por PCR a tiempo real: CD36 (cociente de array 2,30, cociente de PCR 1,67); MNDA (cociente de array 2,312 cociente de PCR 1,79). En cambio, la expresión de guanine nucleotide binding protein α 13 (GNA13), nuclear receptor subfamily 4, group A, member 3 (NOR1), e IL1R2, que resultó incrementada por el tratamiento según los resultados de los arrays, no resultó zsignificativamente modificada cuando se analizó por PCR a tiempo real.
DiscusiónLa patogenia y las anomalías genéticas que se asocian a la HFC no se conocen completamente. En este estudio, se examinó el perfil de expresión génica en monocitos de individuos control sanos normolipémicos y de pacientes afectados por HFC, antes y después de un tratamiento de 4 semanas con atorvastatina.
Aparte de las diferencias en el perfil lipídico, y pese a una adiposidad similar, los pacientes HFC mostraron valores elevados de AGL y reducidos de adiponectina, en comparación con los CT (tabla 2). Estos resultados coinciden con los de Van der Vleuten et al12, que muestran que los valores de adiponectina están reducidos en pacientes HFC, independientemente de la adiposidad, y sugieren un papel del tejido adiposo en la patofisiología de esta enfermedad4. Los valores de proteína C reactiva se hallan incrementados de forma similar en pacientes HFC y en el grupo control, hecho que se podría explicar por la asociación positiva entre el sobrepeso/obesidad y las concentraciones de PCR13. El tratamiento con atorvastatina redujo las concentraciones de lípidos plasmáticos, así como los valores de proteína C reactiva y de otras moléculas inflamatorias (tabla 2), efecto que se relaciona con las propiedades antiinflamatorias de las estatinas14.
En cuanto al perfil de expresión génica, el análisis muestra la expresión diferencial de 82 genes en el grupo HFC en comparación con el grupo CT, y de 88 genes en el grupo HFC tras el tratamiento con atorvastatina en comparación con la situación basal. Muchos de los genes que se hallaron diferencialmente expresados están relacionados con funciones clave en el macrófago y el desarrollo de aterosclerosis, como la respuesta inflamatoria e inmunitaria, el control de la composición de la matriz extracelular (ECM) y el metabolismo lipídico.
Uno de los genes cuya expresión resultó más reprimida en los monocitos de los pacientes con HFC fue MNDA (fig. 1), miembro de la familia de proteínas inducibles por interferón p200, que se encuentra implicada en la regulación del crecimiento y la diferenciación celular15. Se ha descrito que proteínas de esta familia interactúan con un elevado número de factores de transcripción proinflamatorios, como por ejemplo AP-1 y NF-κB16. Resulta interesante que MNDA fue también uno de los genes más reprimidos en un estudio de expresión génica en macrófagos expuestos a LDL oxidadas17. El significado biológico de estos cambios todavía se desconoce, pero dado que MNDA promueve la supervivencia celular, la represión de este gen en la formación de la célula espumosa podría acelerar la muerte de estas células18, uno de los eventos clave en el desarrollo de la placa de ateroma.
 y reacción en cadena de la polimerasa a tiempo real (Taqman) (gris). Los valores se expresan en porcentaje de incremento o reducción en la expresión génica en comparación con su correspondiente control.")
Validación de los genes diferencialmente expresados en las muestras de hiperlipemia familiar combinada comparadas con los controles. Comparación de los resultados obtenidos mediante el array de Affymetrix (negro) y reacción en cadena de la polimerasa a tiempo real (Taqman) (gris). Los valores se expresan en porcentaje de incremento o reducción en la expresión génica en comparación con su correspondiente control.
El entorno proinflamatorio presente en la HFC podría afectar a la expresión de diversos genes que responden a este estímulo, o bien que se encuentran relacionados con la cascada inflamatoria. La interleucina (IL) 1 es una citocina multifuncional, cuya vía de señalización se inicia con la unión al receptor tipo 1 de IL-1 (IL1R1). La señalización mediante IL-1 activa la síntesis de sustancias vasoactivas, factores de crecimiento y citocinas; esta vía proinflamatoria se regula mediante el receptor de tipo 2 (IL1R2), un receptor decoy que no transduce la señal, y que es la forma predominante de receptor de IL-1 en monocitos19. Nuestros resultados indican una disminución de la expresión de IL1R2 en monocitos de pacientes con HFC (fig. 1), lo que facilitaría la señalización a través de IL1R1 y la progresión de la cascada inflamatoria. La represión de IL1R2 podría ser consecuencia de la inflamación característica de la HFC, ya que se ha observado que la transcripción de IL1R2 disminuye debido a señales proinflamatorias19.
En contraste con la represión de los genes mencionados anteriormente, los monocitos de los pacientes con HFC mostraron un incremento en la expresión de ALCAM, LRIG1, y TNFRSF25 (o DR3) (fig. 1). ALCAM pertenece a la superfamilia de las inmunoglobulinas, actúa como molécula de adhesión y se encuentra relacionado con la respuesta inflamatoria e inmunitaria20. El incremento de la expresión de ALCAM observado en nuestro estudio puede atribuirse también al entorno inflamatorio en el que se hallan los monocitos en la HFC, ya que de forma similar se ha descrito un aumento en la expresión de este gen en células del linaje monocítico en pacientes con artritis reumatoide21.Por otra parte, el aumento en la expresión de ALCAM puede estar relacionado con la formación de lesiones ateroscleróticas, ya que se ha observado un aumento en la expresión de este gen en ratones knockout para apo E20.
LRIG1 modula la transducción de señal mediada por receptores de factores de crecimiento; de forma específica, LRIG1 actúa como un regulador negativo de la familia de receptores tirosina-cinasa ErbB, entre los cuales se incluye el receptor del factor de crecimiento epidérmico (EGFR)22. La quimiotaxis de monocitos y macrófagos es una de las etapas clave en el desarrollo de aterosclerosis, y está mediada, entre otros factores, por EGFR23. La activación de EGFR genera en la célula múltiples mecanismos de atenuación que sirven para controlar la amplitud y la duración de la señal. Uno de éstos, denominado atenuación tardía, consiste en la inducción transcripcional de LRIG1, que conlleva la ubiquitinización y posterior degradación de EGFR24. Si se considera el papel de este receptor en la aterosclerosis23, el incremento de la expresión de LRIG1 en los monocitos de pacientes con HFC podría ser un mecanismo adaptativo para bloquear un exceso de señalización por EGF.
El miembro 25 de la superfamilia del receptor del TNF también se denomina death receptor 3 (DR3) por su implicación en la inducción de la apoptosis25. La expresión de DR3 se encuentra incrementada en los monocitos de pacientes con HFC en relación con los del grupo control (fig. 1). A parte de su papel en la apoptosis, la activación de DR3 estimula el factor proinflamatorio NF-κβ26 y la expresión de citocinas y quimiocinas proinflamatorias, así como de metaloproteinasas de matriz27, lo que indica su implicación en la aterogenia. De hecho, Kang et al28 demostraron la expresión de DR3 en placas de ateroma, colocalizando con macrófagos y células espumosas. Estos autores también observaron que monocitos de sangre periférica expresaban valores bajos de DR3, pero la expresión se inducía después de un estímulo proinflamatorio28, situación que también se produce en los monocitos en la HFC, y que podría explicar nuestros resultados.
Otros genes diferencialmente expresados en las muestras de pacientes HFC en comparación con el grupo control están relacionados con la composición y la remodelación de la ECM. La fibronectina (FN) es uno de los principales componentes de la ECM29, y syndecan-2 (SDC2) es un proteoglucano heparán-sulfato que participa en los procesos de adhesión célula-célula y célula-matriz30 y que interactúa y se une a la FN31. Ambos genes se hallan sobreexpresados en los monocitos de los pacientes HFC (fig. 1). La FN producida localmente por las células en el entorno de la lesión puede depositarse en la ECM y estimular la adhesión de monocitos32. El incremento de la expresión de SDC2 podría potenciar la capacidad adhesiva de estas células a la pared vascular33. Así pues, el incremento observado en ambos genes en nuestro estudio, contribuiría al desarrollo de aterosclerosis en la HFC. Otro de los genes que se halla modificado en monocitos de pacientes con HFC, es tissue factor pathway inhibitor-2 (TFPI2), un inhibidor de las serín-proteasas relacionado con la remodelación de la ECM (fig. 1). Los datos sobre la expresión de TFPI2 en lesiones ateroscleróticas son contradictorios, puesto que se ha descrito tanto sobreexpresión34 como represión35. El incremento hallado en nuestro estudio podría tener implicaciones en el desarrollo y la estabilidad de la placa, ya que TFPI2 inhibe la plasmina y de forma directa o indirecta produce la inactivación de metaloproteinasas34.
Algunos genes relacionados con el metabolismo lipídico también resultaron diferencialmente expresados en los monocitos de pacientes HFC. CD36, un receptor scavenger que modula la captación de lipoproteínas modificadas y de células apoptóticas en monocitos y macrófagos36, resultó reprimido casi un 50% (fig. 1). Diversos estudios in vitro han demostrado que la exposición a LDL modificadas incrementa la expresión de CD36 en macrófagos37,38, y que la expresión de CD36 es elevada en macrófagos cargados de lípidos en las placas de ateroma humanas39,40. En nuestro estudio, se examinó la expresión de CD36 en monocitos circulantes, no en macrófagos totalmente diferenciados y cargados de lípidos, hecho que quizá podría explicar la diferencia con los estudios anteriores. En los escasos trabajos en los que se ha evaluado la expresión de ARNm de CD36 en monocitos circulantes de pacientes, no se han observado incrementos en condiciones proaterogénicas41. Por otro lado, diversos estudios han mostrado que la expresión de CD36 en monocitos/macrófagos se encuentra reprimida por numerosos mediadores inflamatorios36. La reducción de la expresión de CD36 observada en nuestro estudio podría, en consecuencia, reflejar el entorno proinflamatorio que rodea a los monocitos circulantes en la HFC.
En cuanto a los resultados obtenidos en pacientes HFC tras el tratamiento con atorvastatina, nuestra atención se centró particularmente en los genes cuya expresión se modificó con el tratamiento en sentido contrario al cambio de expresión observado en la comparación de HFC con los individuos CT. Así, la represión de CD36 y MNDA revirtió después del tratamiento (fig. 2). Del mismo modo, TFPI2, LRIG1 y DR3, cuya expresión estaba aumentada en los pacientes HFC al inicio del tratamiento, resultaron reprimidos después del tratamiento (fig. 2). Teniendo en cuenta que la modificación en la expresión génica posiblemente se halle relacionada con el fenotipo inflamatorio de los pacientes HFC no tratados, la reversión hacia los valores control podría deberse a los efectos antiinflamatorios de la atorvastatina, que se observan por la reducción de los valores de proteína C reactiva después del tratamiento (tabla 2).
 y reacción en cadena de la polimerasa a tiempo real (Taqman) (gris). Los valores se expresan en porcentaje de incremento o reducción en la expresión génica en comparación con su correspondiente control.")
Validación de los genes diferencialmente expresados en las muestras de hiperlipemia familiar combinada después del tratamiento con atorvastatina comparadas con la situación basal. Comparación de los resultados obtenidos mediante el array de Affymetrix (negro) y reacción en cadena de la polimerasa a tiempo real (Taqman) (gris). Los valores se expresan en porcentaje de incremento o reducción en la expresión génica en comparación con su correspondiente control.
No obstante, no todos los cambios en la expresión génica se revirtieron con el tratamiento. El tratamiento con atorvastatina tampoco normalizó los valores bajos de adiponectina ni los valores elevados de AGL. Por otra parte, las concentraciones de triglicéridos plasmáticos se redujeron, pero no llegaron a alcanzarse los valores del grupo control (tabla 2). Estos resultados indican que el tratamiento con atorvastatina no es capaz de corregir ciertas alteraciones de la HFC asociadas a anomalías metabólicas del tejido adiposo. Es posible que algunos de los cambios en la expresión génica observados en nuestro estudio sean consecuencia directa o indirecta de estos efectos metabólicos que el tratamiento con atorvastatina no corrige. Ello explicaría que el tratamiento con atorvastatina, a través de sus efectos antiinflamatorios, sea capaz de revertir o corregir en parte algunos de los cambios en la expresión génica observados en la HFC, mientras que otros genes cuyos cambios en la expresión podrían reflejar anomalías del tejido adiposo no resulten modificados por el tratamiento.
En resumen, nuestros resultados muestran diferencias en la expresión génica de monocitos de pacientes con HFC en comparación con los del grupo CT, algunas de las cuales modula el tratamiento con atorvastatina. El entorno inflamatorio local, y también las anomalías metabólicas en el tejido adiposo, podrían relacionarse con muchos de los cambios de expresión observados en la HFC. El presente estudio ha demostrado que los análisis de expresión génica son útiles a la hora de profundizar en una enfermedad compleja como es el caso de la HFC. Sin embargo, a partir de nuestros resultados, basados en una muestra relativamente pequeña de pacientes e individuos CT, no podemos saber si los cambios observados son respuesta a defectos genéticos primarios, respuestas adaptativas, o ambas cosas.
Queremos agradecer a los pacientes y al grupo control su colaboración y participación en este estudio.
Este estudio ha sido parcialmente subvencionado con fondos de la Fundació Privada Catalana de Nutrició i Lípids, Fundación Ramón Areces, CICYT (SAF2004-03045), Unión Europea FEDER, FIS (RTIC C01/01 y G03/181). Una comunicación referente a esta línea de trabajo fue presentada en el XIX Congreso Nacional de la SEA (Santander, 2006) y galardonada con una mención especial.
Los CIBER Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM) y Fisiopatología de la Obesidad y Nutrición (CIBERBN) son iniciativas del Instituto de Salud Carlos III.