




Postprandial lipemia has been associated with cardiovascular disease. The current pathophysiological concept is that postprandial remnant lipoproteins migrate into the subendothelial space and that remnants activate circulating leukocytes and endothelial cells. Activated monocytes adhere to endothelial adhesion molecules, facilitating subendothelial migration of monocytes. These cells differentiate into macrophages, with the risk of foam cell formation, due to uptake of remnants and modified lipoproteins. Evidence is emerging that specific interventions may reduce the atherogenic postprandial inflammation. Fruits rich in polyphenols, virgin olive oil, carotenoids and exercise have recently been found to reduce postprandial inflammation. Pharmaceutical interventions with fibrates or statins not only improve the overall lipid profile, but reduce postprandial inflammation as well. This review will deal with the current concept of postprandial inflammation in relation to the development of atherosclerosis and potential interventions to reduce postprandial inflammation.
La lipidemia posprandial está relacionada con la enfermedad cardiovascular. El concepto patofisiológico actual es que las partículas remanentes traspasan el endotelio, activan los leucocitos y las células endoteliales. Los monocitos activados se adhieren a la pared endotelial por mediación de moléculas de adhesión, facilitando así la migración de los monocitos al espacio subendotelial. Estas células se transforman en macrófagos, convirtiéndose definitivamente en células espumosas después de haber internalizado las partículas remanentes y otras lipoproteínas modificadas. Recientes estudios sugieren que existen intervenciones efectivas para modular la inflamación posprandial, y de esta forma rebajar el riesgo cardiovascular. Frutas ricas en polifenoles, aceite de oliva virgen, el caroteno y el ejercicio son ejemplos que han demostrado una reducción de la inflamación posprandial. El tratamiento con estatinas y fibratos no solo mejora el perfil lipídico, sino que también rebaja la lipidemia posprandial. Esta revisión describe los recientes conceptos de la inflamación posprandial relacionada con la generación de ateroesclerosis y también trata las intervenciones que pueden influir positivamente en la inflamación posprandial.
When thinking of lipids in association with cardiovascular disease (CVD), the relationship between low-density lipoprotein cholesterol (LDL-C) and atherosclerosis is usually the first one that comes to mind. This is due to the great number of papers published during the last two decades addressing the effect of LDL-C reduction on incident CVD.1
However, atherosclerosis is a multifactorial disease and elevated LDL-C is just one of the many lipid risk factors involved.2 All apolipoprotein (apo) B containing lipoproteins, which include chylomicrons, chylomicron remnants, very-low density lipoproteins (VLDL), intermediate-density lipoproteins (IDL) and LDL, are atherogenic. Increased fasting and non-fasting triglycerides, high plasma apo B concentrations and increased remnant cholesterol, together with elevated lipoprotein(a) and low high-density lipoprotein cholesterol (HDL-C) concentrations, are now recognized as independent risk factors beyond LDL-C.3–5 Elevated triglycerides and remnant cholesterol levels are associated with postprandial lipemia, and most investigators in the field acknowledge the relevance of postprandial hyperlipidemia as a contributing, or even independent, risk factor for atherosclerosis.6 The usual concept in this respect is that the generation of postprandial remnant particles, which can migrate into the subendothelial space and induce a local inflammatory process ultimately leading to foam cell formation, is responsible for vascular deterioration and plaque formation.7 Recent work from different laboratories has expanded this hypothesis to the widely accepted inflammatory pathogenesis of the atherosclerotic plaque.7–9 Novel data suggest that remnants may interact with circulating inflammatory cells, such as neutrophils and monocytes, and with endothelial cells. This interaction induces the generation of chemokines and oxidative stress, causing an intravascular inflammatory response preceding the subendothelial reaction described earlier.8,10–12
In order to improve our understanding of this intravascular inflammation, it is important to comprehend the processes involved in intravascular remodeling of the triglyceride-rich lipoproteins (TRLs).10,13 This review will address the current knowledge about postprandial lipemia and inflammation, and it will give an overview of lifestyle changes and pharmacological interventions.
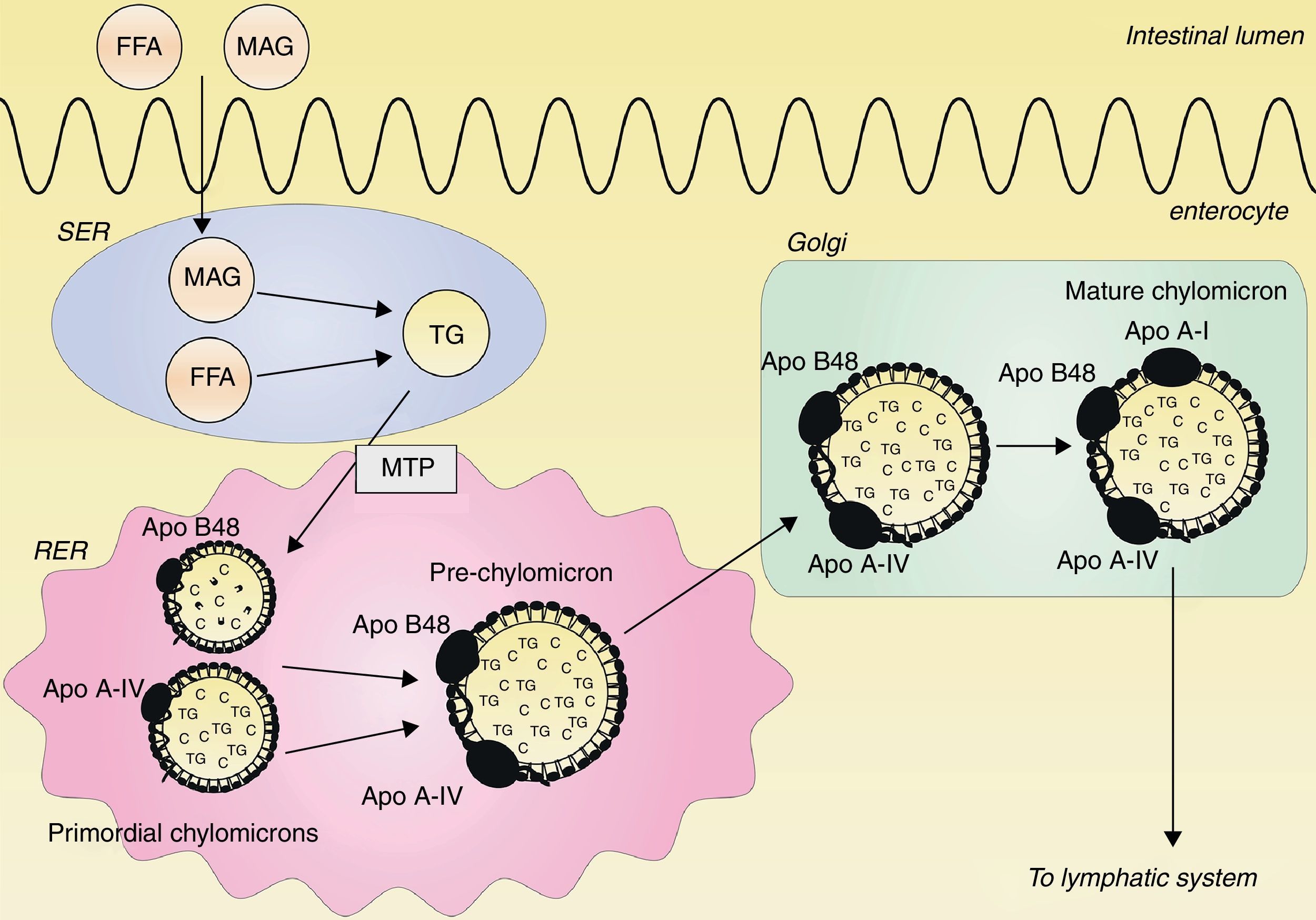
Classical concept of postprandial lipemia: chylomicron synthesis and secretionDietary lipids and fat-soluble vitamins are transported from the intestine to the blood by chylomicrons. Chylomicrons are the largest TRLs, and they contain triglycerides, cholesterol, phospholipids and proteins, with apo B48 as the structural protein.14 Ingested triglycerides are digested into free fatty acids (FFAs) and 2-monoacylglycerol (MAG) by pancreatic lipase. FFAs and MAG are absorbed from the intestinal lumen and are carried to the endoplasmatic reticulum (ER), where they are resynthesized to triglycerides.15–17 Within the ER, a prechylomicron is formed from two different primordial chylomicrons. One primordial chylomicron consists of phospholipids, cholesterol and one apo B48 molecule, and is chaperoned by the microsomal transfer protein (MTP) complex.18 The other primordial chylomicron contains cholesterol esters, triglycerides and a single apo A-IV molecule.19 The prechylomicron is then transported to the Golgi, where apo A-I is attached and a mature chylomicron is formed.19–21 This mature chylomicron is secreted into the lymphatic system.21 This process is illustrated in Fig. 1.
 and 2-monoacylglycerol (MAG) are absorbed from the intestinal lumen and carried to the smooth endoplasmatic reticulum (SER), where they are synthesized to triglycerides (TG). In the rough endoplasmatic reticulum (RER), two primordial chylomicrons are merged to form a prechylomicron. One primordial chylomicron contains a dense lipid core with phospholipids and cholesterol (C) together with one apo B48 molecule, chaperoned by the microsomal transfer protein (MTP) complex. The other primordial chylomicron consists of a large particle with cholesterol esters and triglycerides together with one apo A-IV molecule. The prechylomicron is transported to the Golgi, where apo A-I is attached to the prechylomicron, resulting in a mature chylomicron. This mature chylomicron is secreted into the lymphatic system.")
Assembly of chylomicrons. Free fatty acids (FFAs) and 2-monoacylglycerol (MAG) are absorbed from the intestinal lumen and carried to the smooth endoplasmatic reticulum (SER), where they are synthesized to triglycerides (TG). In the rough endoplasmatic reticulum (RER), two primordial chylomicrons are merged to form a prechylomicron. One primordial chylomicron contains a dense lipid core with phospholipids and cholesterol (C) together with one apo B48 molecule, chaperoned by the microsomal transfer protein (MTP) complex. The other primordial chylomicron consists of a large particle with cholesterol esters and triglycerides together with one apo A-IV molecule. The prechylomicron is transported to the Golgi, where apo A-I is attached to the prechylomicron, resulting in a mature chylomicron. This mature chylomicron is secreted into the lymphatic system.
In the circulation, chylomicrons are hydrolyzed by lipoprotein lipase (LPL), which converts triglycerides into glycerol and fatty acids,22 resulting in the formation of cholesterol-dense lipoprotein remnants, which are taken up by the liver.23,24 Recently, the important role in the hydrolysis of triglycerides of glycosylphosphatidylinositol-anchored high-density lipoprotein binding protein 1 (GPIHBP1) has been identified. This protein serves as a binding site for LPL to the endothelium, and thus provides a docking platform for chylomicrons and VLDL, facilitating the triglyceride lipolysis from these lipoproteins.25,26 The dietary FFAs that are taken up by the liver, may be re-assembled and returned to the blood in VLDL.27 Alternatively, lipolysis of these TRLs attached to the endothelium in the adipose tissue will cause influx of FFAs into adipocytes and re-esterification to triglycerides. Finally, released FFAs may also be taken up by myocytes and enter the fatty acid oxidation pathway. Recent in vivo work demonstrated that only 5% of circulating FFAs are diverted to VLDL-triglyceride, independent of the prandial situation.28
The endothelium is continuously challenged by atherogenic lipoprotein remnants, and since chylomicrons and VLDL share the same metabolic pathway and the availability of the enzymes involved is limited, competition at the level of several enzymes may occur, resulting in accumulation of TRLs.29,30 This competition is most likely to occur when already fasting hypertriglyceridemia is present, which is characterized by increased levels of circulating remnants.31 Potential sites of competition include not only LPL, but also hepatic lipase, another enzyme involved in the hydrolysis of the triglyceride in TRLs,32 and binding of remnants to cell surface receptors which facilitate the clearance of TRL remnant particles, such as the LDL receptor, low-density lipoprotein receptor-related protein 1 (LRP-1) and heparan sulfate proteoglycans.25
Postprandial lipemia and atherosclerosisSeveral large epidemiological studies have established the association between postprandial lipemia and CVD. The Copenhagen City Heart Study demonstrated that elevated non-fasting triglycerides levels are associated with an increased risk of myocardial infarction and ischemic heart disease.33 In addition, a meta-analysis of 29 prospective studies has shown that non-fasting triglycerides are associated with risk of fatal and nonfatal coronary events.3
The relationship between triglycerides and CVD may be caused by its close relationship with levels of remnant cholesterol. Increased non-fasting remnant cholesterol levels were causally associated with CVD using a Mendelian randomization study design.34 Every 1mmol/l increase in serum remnant cholesterol resulted in a 2.8 fold increased risk of incident ischemic heart disease, which was independent from decreased HDL-C levels. However, pleiotropic effects of different genetic variants could not be excluded in this epidemiological study, making experimental and interventional studies necessary to confirm this pathophysiological model.
Smaller studies have elegantly demonstrated the association between postprandial lipemia and carotid intima media thickness (IMT). In 156 healthy subjects, the IMT was independently associated with TRLs, together with small dense LDL.35 Recently, it was demonstrated that serum apo B48, which reflects the number of circulating chylomicrons, correlated best with IMT in patients treated with statins.31
However, there are situations when elevated triglycerides are less strongly related to atherosclerosis, such as in the case of familial hypertriglyceridemia (FTHG).36 This is most likely due to the fact that this disorder, in contrast to familial combined hyperlipidemia, is not accompanied by an increased number of circulating lipid particles. Low plasma apo B levels in FHTG reflect a low number of atherogenic lipoproteins. In this respect, it should be mentioned that plasma apo B levels are also important determinants of postprandial lipemia.37 Significant correlations with postprandial lipemia have also been reported for many other factors, including plasma apo A-IV, apo A-V, apo C-III, apo E, LPL, cholesteryl ester transfer protein, hepatic lipase, complement component 3 (C3) and insulin resistance.38–43
Interaction between postprandial lipoproteins and cellular inflammationMany markers of inflammation, such as leukocyte count, C-reactive protein (CRP) and complement component 3 (C3), have been associated with CVD.41,44–49 A large epidemiological study recently suggested that increased non-fasting remnant cholesterol is causally associated with inflammation in atherosclerosis.50 In contrast, LDL-C was associated with atherosclerosis without inflammation.50 Furthermore, hypertriglyceridemia has been associated with increased levels of high-sensitivity C-reactive protein.51
The activation of leukocytes has been proposed to be an important factor in the development of atherosclerosis.52–54In vitro and in vivo studies have demonstrated that TRLs are able to induce leukocyte activation.55–59 Several animal studies have underlined the association between TRLs and postprandial inflammation.60,61 In mice expressing human APOE2, which is associated with a decreased clearance of TRLs, postprandial inflammation was higher than in mice expressing APOE3.60 Another study in mice demonstrated that VLDL-receptor deficiency, resulting in reduced uptake of remnant particles by adipocytes, leads to decreased inflammation in adipocytes.61
Postprandial leukocyte activation after ingestion of a high-fat meal has also been confirmed in experimental studies with humans.55,62 Van Oostrom et al. have shown that in the postprandial state neutrophil counts increase, with concomitant production of IL-6 and IL-8 and oxidative stress, which resulted in endothelial dysfunction reflected by impaired flow mediated dilatation.67,78 Recently, it was demonstrated that postprandial activated monocytes showed increased adhesion to VCAM-1.62 A study with 45 patients who underwent carotid endarteriectomy revealed that the cholesterol content of TRLs was highly associated with macrophage content in plaques, illustrating the pathophysiological importance of postprandial lipemia and its associated cellular inflammation.79
The mechanism of this leukocyte activation in the circulation is now being disclosed. One of the potential explanations is that pro-inflammatory lipoproteins bind to circulating leukocytes, causing a state of activation.9,11 We have shown that apo B is present on the cellular membrane of neutrophils and monocytes, and that postprandial leukocytes transport dietary fatty acids.55 Others have demonstrated that human leukocytes are also able to internalize remnants in the circulation.63,64 Finally, leukocytes of patients with CVD have been found to have a higher lipid content than those of healthy subjects.65 This internalization of TRLs by leukocytes may be mediated not only by the LDL-receptor, but also by LRP-1, the apo B48 receptor and other yet unidentified receptors.62,63,66 Collectively, these data suggest that direct activation of leukocytes may occur in the circulation by interaction with postprandial lipoproteins.
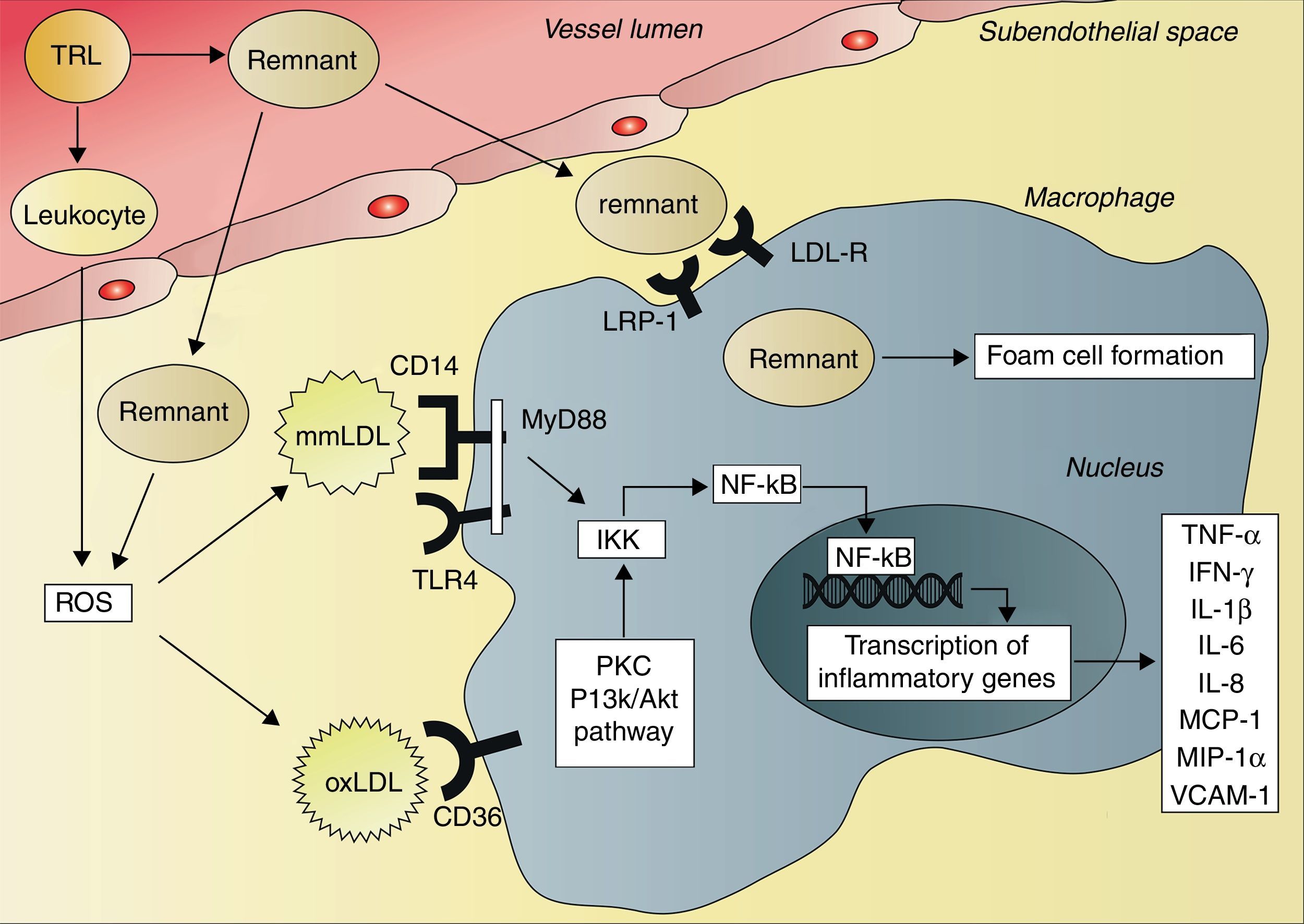
The consequence of these interactions is a situation of systemic inflammation. After ingestion of a high-fat meal, resulting in activation of neutrophils, the production of reactive oxygen species (ROS) increases.67 ROS can initiate lipid peroxidation and thus the formation of minimally modified LDL (mmLDL) and extensively oxidized LDL (oxLDL).68 This postprandial oxidative modification of LDL leads to transcription of inflammatory genes in various cell types, including macrophages and endothelial cells, mediated by the activation of the transcription factor nuclear factor kappaB (NF-κB).69,70 OxLDL and chylomicron remnants are taken up by macrophages, activating the protein kinase C and phosphatidylinositol 3-kinase (PI3K)/Akt pathways.70,71 This results in activation of the IκB kinase complex (IKK), leading to phosphorylation of the N-terminal of IκB proteins and consequently activation of NF-κB.72,73 In contrast to oxLDL and chylomicron remnants, mmLDL is not internalized by macrophages. However, mmLDL is recognized by CD14 on macrophages, which then associates with the toll-like receptor-4 (TRL4), inducing a pro-inflammatory signaling pathway.74 This pathway involves activation of the adapter protein myeloid differentiation primary response gene 88 (MyD88) and subsequently activation of IKK and NF-κB.72,73 Activation of NF-κB leads to an increased expression of genes that encode cytokines, such as tumor necrosis factor-alpha (TNF-α), interferon-gamma (IFN-γ), interleukin-1 beta (IL-1β), IL-6 and IL-8, and different chemokines, such as monocyte chemotactic protein-1 (MCP-1) and macrophage inflammatory protein-1 alpha (MIP-1α), and also cellular adhesion molecules, such as soluble vascular cell adhesion molecule 1 (VCAM-1)62,67,69,75–77 (Fig. 2). This inflammatory cascade may therefore explain the systemic postprandial inflammation and endothelial dysfunction described earlier.
 enter the subendothelial space, where they can be taken up by macrophages via the low-density lipoprotein receptor (LDL-R), the LDL receptor-related protein 1 (LRP-1) and other receptors, resulting in foam cell formation. TRLs also activate circulating leukocytes, leading to the production of reactive oxygen species (ROS). In addition, subendothelial remnants itself induce production of ROS. ROS initiate lipid peroxidation and formation of minimally modified low-density lipoprotein (mmLDL) and extensively modified low-density lipoprotein (oxLDL). The CD14 receptor on macrophages recognizes mmLDL, and CD14 then associates with toll-like receptor 4 (TRL4). This leads to activation of myeloid differentiation primary response gene 88 (MyD88), IκB kinase complex (IKK) and nuclear factor kappaB (NF-κB). OxLDL is taken up by macrophages via the CD36 receptor, leading to activation of protein kinase C (PKC) and phosphatidylinositol 3-kinase (PI3K)/Akt pathways, resulting in activation of IKK and NF-κB. Activation of NF-κB leads to increased transcription of tumor necrosis factor-alpha (TNF-α), interferon-gamma (IFN-γ), interleukin-1 beta (IL-1β), IL-6, IL-8, monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein-1 alpha (MIP-1α) and vascular cell adhesion molecule 1 (VCAM-1).")
Activation of cytokine production by triglyceride-rich lipoproteins and remnants. The remnants of triglyceride-rich lipoproteins (TRLs) enter the subendothelial space, where they can be taken up by macrophages via the low-density lipoprotein receptor (LDL-R), the LDL receptor-related protein 1 (LRP-1) and other receptors, resulting in foam cell formation. TRLs also activate circulating leukocytes, leading to the production of reactive oxygen species (ROS). In addition, subendothelial remnants itself induce production of ROS. ROS initiate lipid peroxidation and formation of minimally modified low-density lipoprotein (mmLDL) and extensively modified low-density lipoprotein (oxLDL). The CD14 receptor on macrophages recognizes mmLDL, and CD14 then associates with toll-like receptor 4 (TRL4). This leads to activation of myeloid differentiation primary response gene 88 (MyD88), IκB kinase complex (IKK) and nuclear factor kappaB (NF-κB). OxLDL is taken up by macrophages via the CD36 receptor, leading to activation of protein kinase C (PKC) and phosphatidylinositol 3-kinase (PI3K)/Akt pathways, resulting in activation of IKK and NF-κB. Activation of NF-κB leads to increased transcription of tumor necrosis factor-alpha (TNF-α), interferon-gamma (IFN-γ), interleukin-1 beta (IL-1β), IL-6, IL-8, monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein-1 alpha (MIP-1α) and vascular cell adhesion molecule 1 (VCAM-1).
The complement system is an important determinant of postprandial lipemia and it has been associated with CVD.47,80 Components of the complement system have been found to co-localize with CRP in early atherosclerotic lesions, indicating that activation of the complement system may be involved in the development of atherosclerotic plaques.81 C3 is one of the strongest determinants of postprandial triglyceridemia, and a postprandial increase in C3 levels has been observed in patients with and without CVD, which was correlated with the postprandial triglyceride response.82In vitro and in vivo studies have demonstrated that chylomicrons are a strong stimulator of the production of C3 by adipocytes.49,83
The biological background of the relationship between the complement system and postprandial lipemia may be the C3/acylation stimulating protein (C3/ASP)-system, an important regulator of fatty acid metabolism in adipose tissue. ASP, which is identical to C3adesArg, is formed when the carboxyl terminus of one of the split products of C3, C3a, is desarginated.84 ASP has several functions: it increases triglyceride storage in adipocytes by stimulating the re-esterification of FFA into triglycerides, it enhances glucose uptake by adipocytes, fibroblasts and muscle cells by increasing the translocation of glucose transporters Glut1, Glut3 and Glut4, and it reduces lipolysis in adipocytes by inhibiting hormone sensitive lipase.84
All these data support the involvement of the complement system as a metabolic regulator of postprandial inflammation. However, taking all these data into account, one should realize that the physiological function of postprandial inflammation is not yet understood. Since postprandial inflammation also occurs in healthy subjects, subtle modulations that are so far unidentified, may be responsible for the predicted relationship with atherosclerosis.
Lifestyle interventions to reduce postprandial inflammationUntil recently there were no known interventions to modulate postprandial inflammation, but evidence from small intervention trials is starting to emerge. Postprandial inflammation may be positively influenced by diet and lifestyle behavior. In a randomized trial with eleven volunteers, the addition of avocado, which is rich in mono-unsaturated FFAs, to a hamburger meal attenuated the postprandial increase of serum IL-6.85 This was probably mediated via reduced activity of the NF-κB pathway, since IκB was preserved in leukocytes. These reductions in postprandial inflammation by avocado were accompanied with reduced vasoconstriction when a hamburger was consumed.85 The type of fat ingested may also influence postprandial inflammation. One study with 20 obese subjects demonstrated that when muffins were prepared with different oils, the postprandial inflammatory response could be altered. Oils rich in phenols, such as virgin olive oil, reduced postprandial inflammation by reducing NF-κB production by leukocytes, together with a reduction in plasma lipopolysaccharide concentrations. On the contrary, sunflower oil exaggerated the postprandial inflammatory response with increased NF-κB, IL-1 and IL-6 mRNA expression levels in leukocytes.86 Addition of antioxidant to oils may have similar effects.86 Recent clinical trials have demonstrated that raspberries and strawberries, rich in polyphenols, attenuate postprandial inflammation.87,88 Consumption of 45 grams of lyophilized black raspberries for four days reduced the postprandial IL-6 response after an oral fat load.87 Consumption of strawberries has been shown to reduce postprandial oxidative stressors in hyperlipidemic men and women.88 Therefore, the use of polyphenols and antioxidants seem to be beneficial in reducing postprandial inflammation, while sunflower oil seems detrimental.
The effects of omega-3 polyunsaturated fatty acids (n3-PUFA) remain controversial. Supplementation with n-3 PUFAs during six months reduced the fasting and postprandial production of several inflammatory parameters, including sICAM, TNF-α and IL-6 when compared to placebo.89 However, a randomized placebo-controlled trial demonstrated that ingestion of n3-PUFA during three weeks lowered fasting and postprandial triglycerides, but postprandial leukocyte activation and sICAM production was not reduced.90
Interestingly, consumption of tomatoes containing carotenoids has been shown to reduce postprandial oxidation of LDL and rise in serum IL-6 levels, despite an increased postprandial lipemia.91 Although regular consumption of red wine has been associated with reduced CVD risk,92 red wine had no effect on inflammation after consumption of a high-fat meal.93 Besides dietary interventions, exercise should also be considered as a potential modulator of postprandial inflammation. Exercise shortly before a high-fat meal blunted the postprandial increase in the expression of CD11a and CD18 on monocytes in healthy volunteers.94 However, exercise did not attenuate the postprandial cytokine production.94,95 More studies are needed to evaluate the effects of exercise on postprandial inflammation.
Pharmaceutical interventions to reduce postprandial inflammationCertain drugs have been found to reduce postprandial inflammation, but it should be noted that most of these drugs also improve the lipid profile. Therefore, it remains unsure whether the reduction in postprandial inflammation is a direct effect from the drug or the result of a reduction in lipoproteins.
Fibrates seem to inhibit NF-κB.75 In two randomized, placebo-controlled trials, fenofibrate treatment reduced the postprandial production of TNF-α, IL-1β, IL-6, MCP-1 and MIP-1α.75,96 It should be noted that while these effects of fibrates suggest protection against atherosclerosis, the clinical data do not fully support this yet, and there is still controversy on the effect of fibrates on the development of atherosclerosis.
Several studies have assessed the effect of statins on postprandial C3 levels and leukocyte activation. High-dose simvastatin has been demonstrated to blunt the postprandial increase in C3 in patients with coronary artery disease.82 Atorvastatin reduced postprandial C3 increment in patients with familial combined hyperlipidemia.49 In another study among patients with coronary artery disease, rosuvastatin treatment reduced migration of neutrophils and endothelial cell activation, but did not reduce postprandial leukocyte activation.97 Thus, some aspects of the postprandial inflammatory response may be modified by statins.
Antidiabetic drugs have also been demonstrated to attenuate postprandial inflammation.98,99 In a randomized placebo-controlled trial, treatment of 31 human immunodeficiency virus-infected patients with rosiglitazone or metformin increased postprandial the activity of paraoxonase (PON)-1, an enzyme with anti-oxidant properties, and reduced postprandial MCP-1 concentrations.99 The anti-inflammatory effect of rosiglitazone was confirmed by another trial, where treatment of 19 patients with type 2 diabetes mellitus with rosiglitazone resulted in decreased fasting plasma peroxides, and increased levels of fasting and postprandial PON-1.98
ConclusionDuring postprandial lipemia, TRLs induce complement activation, cytokine production, leukocyte activation and oxidative stress, contributing to endothelial dysfunction and the development of atherosclerosis. The physiological relevance of postprandial inflammation is yet unknown, but a large body of evidence suggests stimulation of pro-atherogenic pathways. Therefore, it is probably beneficial to reduce postprandial inflammation by specific lifestyle and pharmaceutical interventions, but definitive data are lacking. Consumption of virgin olive oil, black raspberries and strawberries, which are all rich in phenols, and carotenoids present in tomatoes, are effective in reducing postprandial inflammation. Exercise also seems to blunt postprandial leukocyte activation. Finally, fibrates and statins not only improve lipid profiles, but also reduce postprandial inflammation.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflict of interestThe authors declare not to have any conflict of interest.