




El pronóstico de la epilepsia está determinado fundamentalmente por la etiología; se asocia en general peor evolución con comienzo precoz de las crisis.
Material y métodosSe revisa nuestra experiencia en epilepsia en niños nacidos después del 1-1-1997 y que presentaron la primera crisis antes de enero de 2008 a los 1-3 meses de edad.
ResultadosSe incluyen 18 casos con el diagnóstico de epilepsia y primera crisis entre 1 y 3 meses de edad. Un caso corresponde al espectro de síndrome de Dravet con la mutación en heterocigosis c829 T>G c277G del gen SCN1A. Cuatro son epilepsias criptogénicas y 13, sintomáticas: 2 errores congénitos del metabolismo (deficiencia de biotinidasa con respuesta a biotina y síndrome de Leigh), 2 de etiología infecciosa y los 9 restantes, encefalopatías prenatales; 9 (50%) tienen un grave retraso psicomotor en la actualidad y 2 fallecieron. En comparación, los casos criptogénicos tuvieron peor evolución.
ConclusionesNuestra experiencia corrobora el mal pronóstico asociado al inicio precoz, entre 1 y 3 meses, de las crisis epilépticas. Dado el amplio abanico etiológico y el pronóstico sombrío, en ausencia de tratamiento específico, es obligada una adecuada estrategia diagnóstico-terapéutica que evite incertidumbres diagnósticas e identifique casos potencialmente tratables como algunos errores congénitos del metabolismo. En este grupo de edad el protocolo de convulsiones de causa no aclarada debe ser el mismo que el de las convulsiones neonatales, incluido el tratamiento con cóctel vitamínico, tras la recogida de muestras biológicas.
The prognosis of epilepsy is basically determined by its aetiology. Early onset of seizures is generally associated with poor progress.
Material and methodsWe review our experience in epilepsy with children born after 1 January 1997 and who had their first seizure between 1 and 3 months of age before January 2008.
ResultsEighteen cases diagnosed with epilepsy and a first seizure between 1 and 3 months of age were included. One case was within the Dravet syndrome spectrum with the c829 T>G c277G heterozygous mutation of the SCN1A gene. Four were cryptogenic epilepsies and thirteen were asymptomatic: 2 were inborn errors of metabolism (biotinidase deficiency with a response to biotin and Leigh's syndrome); 2 were of infectious origin and the remaining nine prenatal encephalopathy. Nine (50%) currently have a severe psychomotor delay and 2 died. The cryptogenic cases had a relatively poor progress.
ConclusionsOur experience corroborates the poor prognosis associated with early onset, between 1 and 3 months, of epileptic seizures. Given the wide aetiological range and the poor prognosis in the absence of specific treatment, an appropriate diagnostic-therapeutic strategy is required to avoid diagnostic uncertainties and can identify potentially treatable cases, such as some inborn errors of metabolism. In this age group, the protocol for convulsions of unknown cause must be the same as that for neonatal convulsions, including treatment with a vitamin cocktail, after collecting biological samples.
En los últimos años se han producido cambios importantes en epileptología que incluyen: la consolidación de los síndromes epilépticos, la confirmación e identificación de epilepsias genéticamente determinadas, la discusión sobre la utilidad o necesidad del tratamiento en determinados casos, la creciente preocupación por la calidad de vida y los aspectos neuropsicológicos del niño epiléptico y por las posibles repercusiones de los tratamientos antiepilépticos, la mejora de los procedimientos diagnósticos, la incorporación de nuevos fármacos y el auge de la cirugía de la epilepsia. Estos avances asocian un cambio en la orientación de la epilepsia y del niño epiléptico, así como una mayor exigencia a los profesionales encargados de su manejo.
El pronóstico de la epilepsia está determinado básicamente por su etiología1,2, por lo que consideramos necesaria la aproximación etiológica a su estudio. Otro factor pronóstico es la edad de inicio de las crisis, y se asocia en general una evolución peor al comienzo precoz de las crisis3,4.
Se revisa nuestra experiencia desde el punto de vista etiológico, electroencefalográfico y evolutivo de los casos de epilepsia de inicio en el grupo de edad de 1 a 3 meses de vida.
Material y métodosSe realiza un estudio retrospectivo de las historias clínicas de los niños nacidos después del 1 de enero de 1997 hasta el 31 de diciembre de 2007, diagnosticados de epilepsia cuya primera crisis epiléptica tuvo lugar entre el mes y los 3 meses de edad y que constan en la base de datos de la Unidad de Neuropediatría del Hospital Universitario Miguel Servet de Zaragoza. En trabajos anteriores, se expuso detalladamente el método empleado en la configuración de las bases de datos utilizadas y los criterios para evaluar los motivos de consulta y establecer los diagnósticos5–9.
Se ha considerado epilepsia cuando se han dado al menos dos crisis epilépticas espontáneas10,11.
El término encefalopatía se ha utilizado obedeciendo a su significado etimológico de padecimiento encefálico, independientemente de su carácter difuso o localizado y de las repercusiones clínicas. Se han considerado encefalopatías posnatales a las secundarias a infecciones del SNC, accidentes y accidentes cerebrovasculares posnatales.
El diagnóstico de encefalopatía prenatal se ha establecido considerando criterios clínicos y/o de neuroimagen. Apoyan el origen prenatal de una encefalopatía datos como los polihidramnios, rasgos dismórficos faciales y malformaciones extraneurológicas concomitantes, y la ausencia de evidencia de noxa perinatal o posnatal. Es diagnóstico de encefalopatía prenatal la identificación por neuroimagen de agenesia de cuerpo calloso, trastornos de la migración neuronal u otras anomalías malformativas.
Ante la ausencia de certeza, se deben clasificar las epilepsias como presumiblemente sintomáticas o criptogénicas, considerándolas así cuando no se han podido clasificar como idiopáticas ni como sintomáticas11.
ResultadosEn el periodo de estudio constan un total de 18 casos de epilepsia que comenzó entre el mes y los 3 meses de vida. Se trata de 13 (72%) epilepsias sintomáticas, 4 (22%) criptogénicas y 1 idiopática correspondiente al espectro del síndrome de Dravet con la mutación en heterocigosis c.829 T>G c277G del gen SCN1A.
En la tabla 1 se muestran las etiologías de las epilepsias sintomáticas.
Etiología de las epilepsias sintomáticas
| Encefalopatías prenatales | 9 (70%) 3 |
| Encefalopatías prenatales sin especificar | 3 |
| Lesión focal prenatal del territorio cerebral media izquierda | 1 |
| Agenesia de cuerpo calloso y malformación hipocámpica bilateral | 1 |
| Esquisencefalia bilateral de labios abiertos, agenesia de cuerpo calloso y displasia cortical difusa | 1 |
| Esquisencefalia bilateral de labios cerrados y agenesia de cuerpo calloso | 1 |
| Holoprosencefalia con displasia cortical difusa | 1 |
| Quiste porencefálico insuflante12 | 1 |
| Errores congénitos del metabolismo | 2 (15%) |
| Deficiencia de biotinidasa | 1 |
| Compatible con síndrome de Leigh* | 1 |
| Encefalopatías posnatales | 2 (15%) |
| Encefalitis viral | 1 |
| Meningitis bacteriana | 1 |
Niña con cuadro clínico-radiológico compatible con síndrome de Leigh: hiperlactacidemia persistente y alteraciones simétricas en núcleos pálidos, putámenes y tálamos evidenciadas en la tomografía computarizada y la resonancia magnética. Antes de las crisis presentaba importantes retraso psicomotor e hipotonía y escasas funciones visuales. Precisó ventilación asistida y perfusión de pentotal para el control de las crisis; falleció en la UCIP a los 7 meses. Los estudios en piel y músculo no fueron concluyentes.
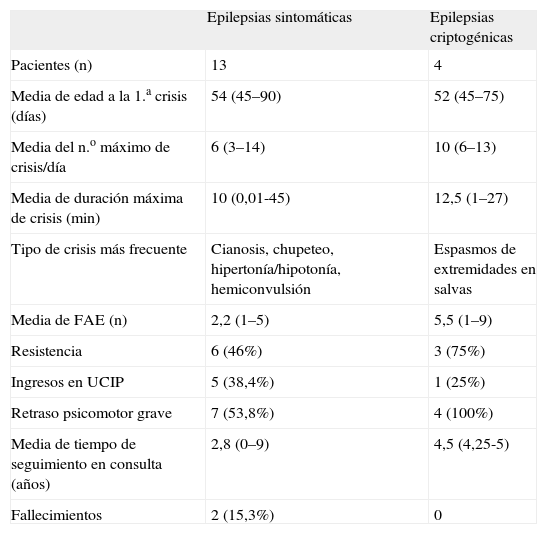
En la tabla 2 se exponen las diferentes variables estudiadas en cuanto a aspectos clínicos y evolutivos de las sintomáticas y las criptogénicas.
Aspectos clínicos y evolutivos de las epilepsias sintomáticas y criptogénicas
| Epilepsias sintomáticas | Epilepsias criptogénicas | |
| Pacientes (n) | 13 | 4 |
| Media de edad a la 1.a crisis (días) | 54 (45–90) | 52 (45–75) |
| Media del n.o máximo de crisis/día | 6 (3–14) | 10 (6–13) |
| Media de duración máxima de crisis (min) | 10 (0,01-45) | 12,5 (1–27) |
| Tipo de crisis más frecuente | Cianosis, chupeteo, hipertonía/hipotonía, hemiconvulsión | Espasmos de extremidades en salvas |
| Media de FAE (n) | 2,2 (1–5) | 5,5 (1–9) |
| Resistencia | 6 (46%) | 3 (75%) |
| Ingresos en UCIP | 5 (38,4%) | 1 (25%) |
| Retraso psicomotor grave | 7 (53,8%) | 4 (100%) |
| Media de tiempo de seguimiento en consulta (años) | 2,8 (0–9) | 4,5 (4,25-5) |
| Fallecimientos | 2 (15,3%) | 0 |
Cuatro de las epilepsias sintomáticas no siguieron controles en nuestro hospital. Los casos que fallecieron corresponden al probable síndrome de Leigh y a la holoprosencefalia.
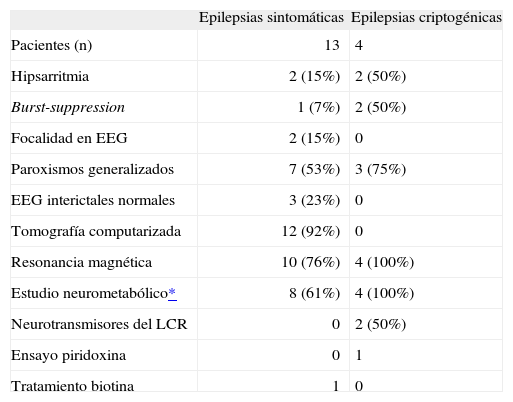
La tabla 3 recoge los patrones electroencefalográficos y los datos de los exámenes complementarios realizados.
Patrones electroencefalográficos y exploraciones complementarias y administración de tratamiento vitamínico
| Epilepsias sintomáticas | Epilepsias criptogénicas | |
| Pacientes (n) | 13 | 4 |
| Hipsarritmia | 2 (15%) | 2 (50%) |
| Burst-suppression | 1 (7%) | 2 (50%) |
| Focalidad en EEG | 2 (15%) | 0 |
| Paroxismos generalizados | 7 (53%) | 3 (75%) |
| EEG interictales normales | 3 (23%) | 0 |
| Tomografía computarizada | 12 (92%) | 0 |
| Resonancia magnética | 10 (76%) | 4 (100%) |
| Estudio neurometabólico* | 8 (61%) | 4 (100%) |
| Neurotransmisores del LCR | 0 | 2 (50%) |
| Ensayo piridoxina | 0 | 1 |
| Tratamiento biotina | 1 | 0 |
Estudio neurometabólico: glucosa, urea, creatinina, ácido úrico, colesterol, albúmina, GOT, GPT, GGT, CPK, iones, equilibrio ácido-base, calcio, amonio, láctico, aminoácidos, homocisteína, beta-OH-butirato, acetoacetato, ácidos grasos libres, triglicéridos, fósforo, fosfatasa alcalina, hormonas tiroideas, cobre, ceruloplasmina, cromatografía de ácidos grasos (AGCML) y realización de CDT (para diagnóstico de los defectos de glucosilación de proteínas). Mucopolisacáridos y ácidos orgánicos en orina. Dry-spot para acilcarnitinas si hay sospecha de alteración del metabolismo intermediario-beta-oxidación y sulfitest en orina si hay hipouricemia comprobada.
Tres casos (16%) tuvieron un patrón electroencefalográfico burst-suppression al inicio, compatible con encefalopatía epiléptica infantil precoz o síndrome de Ohtahara (SO): 1 caso de deficiencia de biotinidasa y 2 de causa no aclarada. Los 2 casos de SO criptogénico presentaron espasmos en salvas y ausencia de contacto visual con hipotonía y tendencia a la somnolencia y un fenotipo morfológico normal. En los 2 casos la resonancia magnética (RM) cerebral y el estudio analítico neurometabólico fueron normales, incluidos neurotransmisores en el líquido cefalorraquídeo en uno de ellos. Los electroencefalogramas (EEG) intercríticos mostraron paroxismos generalizados con la evolución. Se introdujeron durante el tiempo de seguimiento 4 y 9 fármacos antiepilépticos, respectivamente, en un caso se probó corticotropina (ACTH) y piridoxina. Los dos tienen una grave afección cognitiva con ausencia de contacto social, y en un caso persisten las crisis epilépticas tras 4 años de seguimiento.
El caso de déficit de biotinidasa comenzó con crisis epilépticas a los 2 meses de vida, consistentes en espasmos de extremidades superiores durante varios segundos hasta seis diarios. El primer EEG mostró una actividad de fondo desorganizada con abundantes brotes de aplanamiento en relación con las sacudidas de extremidades. A la exploración llamaba la atención el fenotipo morfológico (facies grande, raíz nasal ancha, labios gruesos y orejas de implantación baja), así como la hipotonía cervicoaxial y la ausencia de contacto visual. La RM cerebral mostró una mielinización muy escasa tanto supratentorial como infratentorial. Los ácidos orgánicos en orina fueron compatibles con deficiencia parcial de biotinidasa que posteriormente se confirmó en sangre. El ácido valproico fue ineficaz, y las crisis desaparecieron a los 2 días de administrar biotina a 20 mg/día, con lo que también se normalizó la actividad electroencefalográfica. Actualmente, con 1 año de vida, el paciente sigue sin crisis, y persiste un moderado retraso psicomotor.
El paciente con el espectro Dravet tuvo la primera crisis epiléptica a la edad de 2 meses, consistente en movimientos clónicos de la extremidad superior derecha. Dos meses después presentó otra crisis de 20 min de duración en el contexto de un síndrome febril. Posteriormente, tuvo varias hemiconvulsiones de hemicuerpos derecho o izquierdo indistintamente seguidas, en varias ocasiones, de paresia poscrítica, y cuando tenía 8 meses ingresó en la UCI por estado convulsivo de más de 1 h de duración, en el contexto de bronquitis con fiebre. Inicialmente se pautó ácido valproico, que fue sustituido por carbamazepina, con aumento del número de crisis hasta siete diarias y aparición de crisis parciales complejas y mioclonías. La RM cerebral fue normal, así como el estudio metabólico y los repetidos EEG. Como antecedentes familiares destaca una probable epilepsia en la infancia de la madre. Las frecuentes crisis con fiebre y procesos infecciosos, así como la larga duración de éstas y el empeoramiento con carbamazepina, llevó, a los 9 meses de edad, al análisis MLPA de SCN1A y secuenciación de SCN1A y GABRG2, en que se observó la mutación en el gen SCN1A. Actualmente, con 2 años, el paciente está en tratamiento con levetiracetam y topiramato y persisten crisis cortas semanales.
De los 18 casos, 2 (11%) fallecieron (los casos de holoprosencefalia y sospecha de síndrome de Leigh) y 4 (22%) no siguen controles en nuestro centro. Nueve (50%) presentan un grave retraso psicomotor en la actualidad. Dos niños (11%) presentaron un retraso psicomotor leve y actualmente están libres de crisis y sólo tienen leves dificultades escolares; no precisan educación especial: el afecto de quiste temporoparietal izquierdo insuflante (6 años, actualmente) y el afecto de lesión focal prenatal en el territorio de la cerebral media izquierdo (10 años, actualmente), ambos probablemente secundarios a accidente cerebrovascular prenatal. El paciente afecto de síndrome de Dravet, actualmente con 2 años, tiene retraso en el lenguaje y déficit de atención y comprensión.
DiscusiónFrecuentemente se debate sobre la inocuidad o nocividad de las crisis convulsivas durante los primeros años de vida. Numerosos estudios apuntan a que la repercusión cognitiva de las epilepsias es tanto mayor cuanto menor sea la edad de inicio de las crisis13. Por otro lado, la mayor parte de las epilepsias en este grupo de edad obedecen a etiologías de muy mal pronóstico en ausencia de tratamiento específico, y la repercusión en el neurodesarrollo está determinada fundamentalmente por dichas etiologías.
Los síndromes epilépticos severos en la infancia se caracterizan básicamente por ser dependientes de la edad y de etiología sintomática o criptogénica, hechos que, junto con su resistencia a los farmacos, los configuran como entidades temidas por su mal pronóstico global, tanto en el control de la epilepsia como en el deterioro que conllevan14. Las encefalopatías epilépticas dependientes de la edad comprenden el SO, el síndrome de West y el síndrome de Lennox-Gastaut.
El SO es la forma más precoz de encefalopatía epiléptica15. Se caracteriza por comienzo muy precoz de las crisis, en el periodo neonatal o durante la lactancia antes de los 3 meses16, crisis en forma de espasmos o contracciones tónicas, resistencia a fármacos, mal pronóstico con importante retraso psicomotor, EEG intercrítico con supresión y salvas y variedad etiológica, aunque para muchos autores el diagnóstico de SO comprende un síndrome neurológico de etiología desconocida17.
El SO es la más resistente de las encefalopatías epilépticas dependientes de la edad y la terapia con ACTH no es tan efectiva como en el síndrome de West15. El pronóstico es muy pobre, con muerte precoz o severas secuelas18. El trazado electroencefalográfico denominado salva-supresión, o burst-suppression, es la expresión de una grave afección de la electrogénesis cerebral19. Aunque este trazado es más propio del neonato y durante los primeros meses de vida, recientemente se ha publicado un caso de persistencia de burst-suppression en una niña de 5 años con síndrome de Ohtahara20.
Destacamos el caso de deficiencia de biotinidasa, con espectacular respuesta clínica y electroencefalográfica a la administración de biotina, mientras que los fármacos antiepilépticos tienen escasa o nula respuesta. Es importante el inicio precoz del tratamiento con biotina ya que el déficit neurológico, una vez establecido, será permanente21.
Otro síndrome epiléptico severo de la infancia es el de Dravet o epilepsia mioclónica severa de la infancia. Se inicia en el primer año de vida y se caracteriza por crisis prolongadas e incluso estados epilépticos, normalidad inicial de los estudios electroencefalográficos, resistencia a todo tratamiento antiepiléptico y mal pronóstico funcional con deterioro cognitivo22. Las crisis suelen coincidir con ascensos térmicos y son episodios convulsivos inicialmente de tipo clónico, generalizados o unilaterales, se repiten a intervalos cortos y pueden seguirse de parálisis poscríticas. Son frecuentes antecedentes familiares de crisis febriles o epilepsia. Claes et al23, en 2001, encontraron una nueva mutación en los canales de sodio (SCN1A) en 7 pacientes, similar a la encontrada en la epilepsia generalizada con crisis febriles plus (EGCF+). Mutaciones en el gen SCN1A se han identificado en la epilepsia generalizada con crisis febriles plus y en la epilepsia mioclónica severa de la infancia. El predominio de crisis epilépticas con fiebre es una característica común a las dos entidades. El síndrome de Dravet podría ser parte del espectro de las EGCF+. Nabbout et al, en 2003, observaron que el 10% de las mutaciones del SCN1A se heredan de progenitores asintomáticos o levemente afectados24 y todavía se desconoce por qué en algunas familias con mutaciones demostradas en SCN1A el fenotipo incluye casos de Dravet y familiares asintomáticos, lo que indica la participación de otros genes implicados.
Destacamos la importancia del diagnóstico precoz de un problema que conlleva tanta angustia por la resistencia y el número y la duración de las crisis, lo que permite evitar incertidumbres y repetidos exámenes complementarios y optimizar el tratamiento, dadas sus peculiaridades terapéuticas.
La carbamazepina25 y la lamotrigina pueden agravar el cuadro, mientras que el valproato, el levetiracetam26 y el topiramato27 pueden ser efectivos y bien tolerados. En nuestro caso, hubo un claro agravamiento con la carbamazepina y la situación es aceptable con levetiracetam y topiramato.
En conclusión, nuestra experiencia es compatible con el mal pronóstico que conlleva la epilepsia de inicio precoz descrito en otras series, especialmente en los casos criptogénicos. Aunque somos conscientes de que la muestra es pequeña, todos nuestros pacientes con epilepsia criptogénica presentaron patrones electroencefalográficos más alterados, mayor resistencia y grave retraso psicomotor. Sin embargo, hay casos que pueden responder a un tratamiento específico, como la deficiencia de biotinidasa. En otros casos, el diagnóstico preciso, además de evitar incertidumbres, permite orientar el tratamiento más adecuado, como el caso diagnosticado de síndrome de Dravet.
Dado el amplio abanico etiológico, las posibilidades terapéuticas y el pronóstico sombrío en ausencia de tratamiento específico, es obligada una adecuada estrategia diagnóstico-terapéutica que identifique casos potencialmente tratables, como algunos errores congénitos del metabolismo. Creemos que en este grupo de edad el protocolo de convulsiones de causa no aclarada debe ser el mismo que el de las convulsiones neonatales, incluido el tratamiento con cóctel vitamínico, tras la recogida de muestras biológicas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

