



La gliomatosis cerebral es un tumor astrocítico difuso que afecta a más de dos lóbulos cerebrales. El tratamiento no está bien definido y el pronóstico es malo.
MétodosEstudio retrospectivo clínico-radiológico de 22 pacientes diagnosticados de gliomatosis cerebral en una unidad de neurooncología.
ResultadosEn un periodo de 6 años, identificamos a 17 varones y 5 mujeres (media de edad, 54 años). Los síntomas iniciales fueron déficit focales sensitivo-motores o visuales (86,4%), crisis epilépticas (36,4%), deterioro cognitivo (27,3%) y cefalea (27,3%); en algunos casos los síntomas semejaban ictus, migraña o encefalitis límbica. Todos los pacientes tenían afectación radiológica bilateral; las regiones más afectadas fueron: temporal (19 pacientes), ganglios basales (18), frontal (17), parietal (17), cuerpo calloso (10) y occipital (9). Los diagnósticos histológicos más frecuentes fueron astrocitoma de grado III (36,4%), astrocitoma de grado II (22,7%) y astrocitoma de grado IV (18,3%). Nueve pacientes fueron diagnosticados en el primer mes del desarrollo de los síntomas; 11, entre el primer mes y 1 año, y 2, después de 1 año. Diecisiete pacientes recibieron quimioterapia, radioterapia o ambas, de los que 12 (70,6%) tuvieron respuesta clínica o radiológica. La media de seguimiento fue 13 meses; el tiempo libre de progresión, 6 meses, y el tiempo de supervivencia, 9,5 meses (15 meses cuando los pacientes recibieron tratamiento); 8 pacientes desarrollaron complicaciones tromboembólicas.
ConclusionesLa gliomatosis cerebral tiene un curso clínico variable. Los pacientes generalmente responden al tratamiento. En este estudio la media de supervivencia de los pacientes tratados es similar a la de las series de glioblastoma multiforme.
Gliomatosis cerebri is a diffuse astrocytic neoplasm that involves more than two lobes of the brain. Treatment is not well defined and the prognosis is considered poor.
MethodsRetrospective analysis of 22 patients with gliomatosis cerebri.
ResultsWe identified 17 men and 5 women (median age 54 years) seen in a Division of Neuro-oncology over a 6 year period. Patients presented with focal sensorimotor or visual deficits (86.4%), seizures (36.4%), cognitive dysfunction (27.3%), or headache (27.3%), suggesting in some cases stroke, migraine, or limbic encephalitis. All patients had bilateral involvement; the regions involved included, temporal (19), basal ganglia (18), frontal (17), parietal (17), corpus callosum (10), and occipital (9). The most frequent pathological findings were grade III astrocytoma (36.4%), grade II astrocytoma (22.7%), and grade IV astrocytoma (18.3%). Nine patients were diagnosed within the first month of symptom development, 11 between the first month and 1 year, and 2 after one year. Seventeen patients received treatment with chemotherapy, radiotherapy or both, and 12 patients (70.6%) had a clinical or radiological response. The median follow-up was 13 months, median progression free survival 6 months, and median survival 9,5 months (15 months if the patients received treatment). Eight patients had thromboembolic events.
ConclusionsGliomatosis cerebri has a variable clinical course. Treatment often results in clinical responses. In this study de median survival of patients who received treatment was similar to that reported in series of glioblastoma multiforme.
La gliomatosis cerebral (GC) es una infiltración cerebral difusa de células gliales neoplásicas que preserva la arquitectura del tejido cerebral. Desde que Nevin1 la describió por primera vez en 1938, se han publicado unos 300 casos, fundamentalmente en estudios retrospectivos2. Las manifestaciones clínicas son inespecíficas, por lo que puede confundirse con otras entidades neurológicas y retrasar su diagnóstico. Los objetivos de este trabajo son describir las características clínicas, el tratamiento y evolución de 22 pacientes con GC.
Pacientes y métodosRevisamos las bases de datos e historias clínicas de los pacientes diagnosticados de GC entre enero de 2003 y septiembre de 2009 en la Unidad de Neurooncología del Hospital de la Universidad de Pennsylvania. El criterio diagnóstico de GC según la OMS es una infiltración neoplásica cerebral difusa de células gliales, que preserva la arquitectura del tejido cerebral normal, afecta a más de dos lóbulos cerebrales y, ocasionalmente, a estructuras infratentoriales o médula espinal3. Definimos la GC primaria como la que aparece sin antecedentes de tumor cerebral, y secundaria, la que aparece a partir de un tumor cerebral previo3. Dividimos la GC primaria en tipo 1, una infiltración difusa sin masa tumoral adyacente, y tipo 2, con una masa tumoral adyacente a la infiltración4. De acuerdo con estos criterios, todos los pacientes tuvieron estudios de resonancia magnética (RM) y confirmación histológica del tumor.
Definimos el tiempo de retraso diagnóstico como el transcurrido desde el síntoma inicial hasta el diagnóstico (el valor 0 representa un retraso menor de 1 mes). El tiempo de seguimiento son los meses entre el diagnóstico y septiembre de 2009 o el fallecimiento. La respuesta terapéutica se define como mejoría clínico-radiológica o mejoría clínica con estabilidad radiológica o estabilidad clínico-radiológica. La falta de respuesta se define como empeoramiento clínico o radiológico. El tiempo libre de progresión se considera el transcurrido desde la administración de un tratamiento hasta la evidencia de progresión.
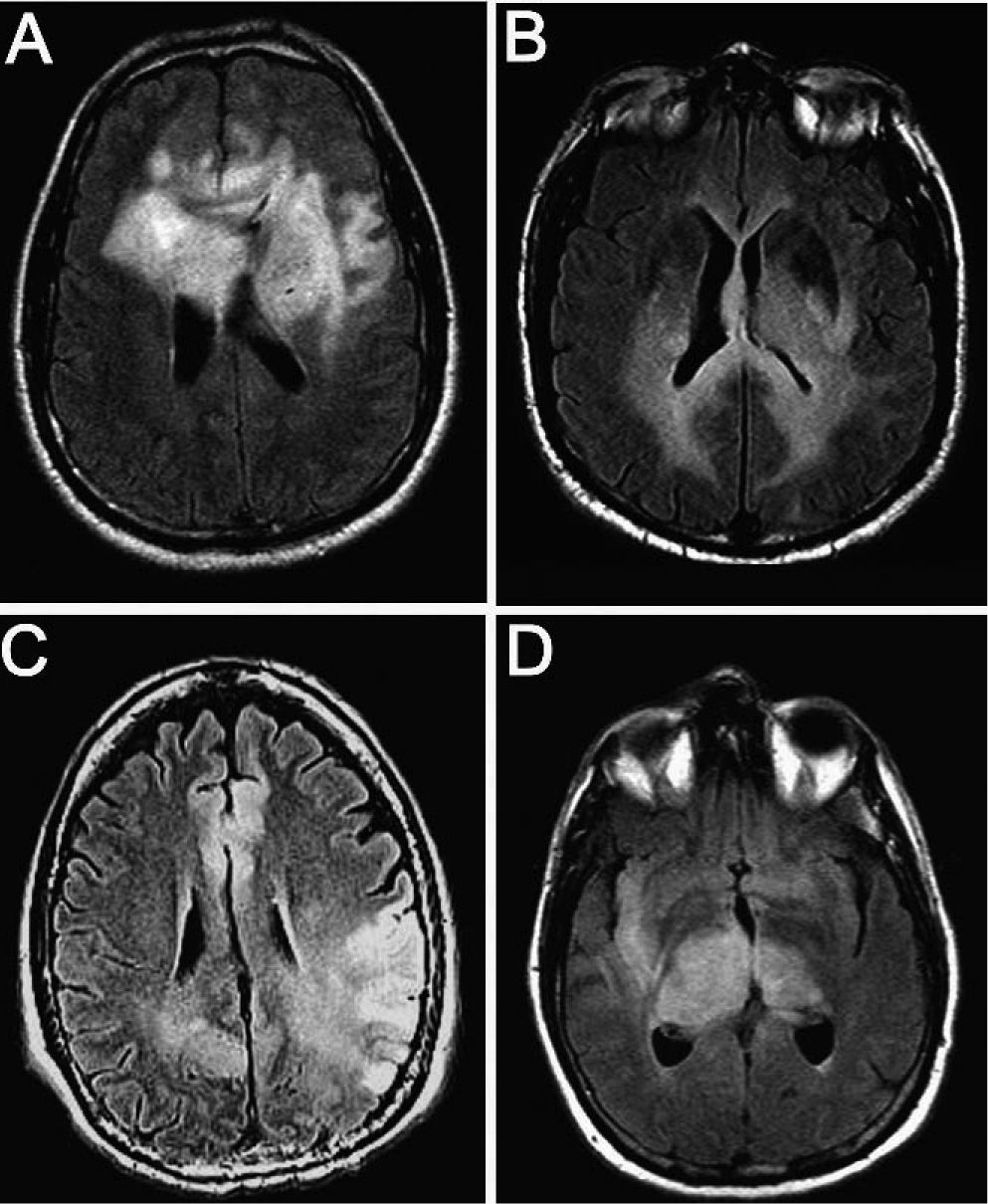
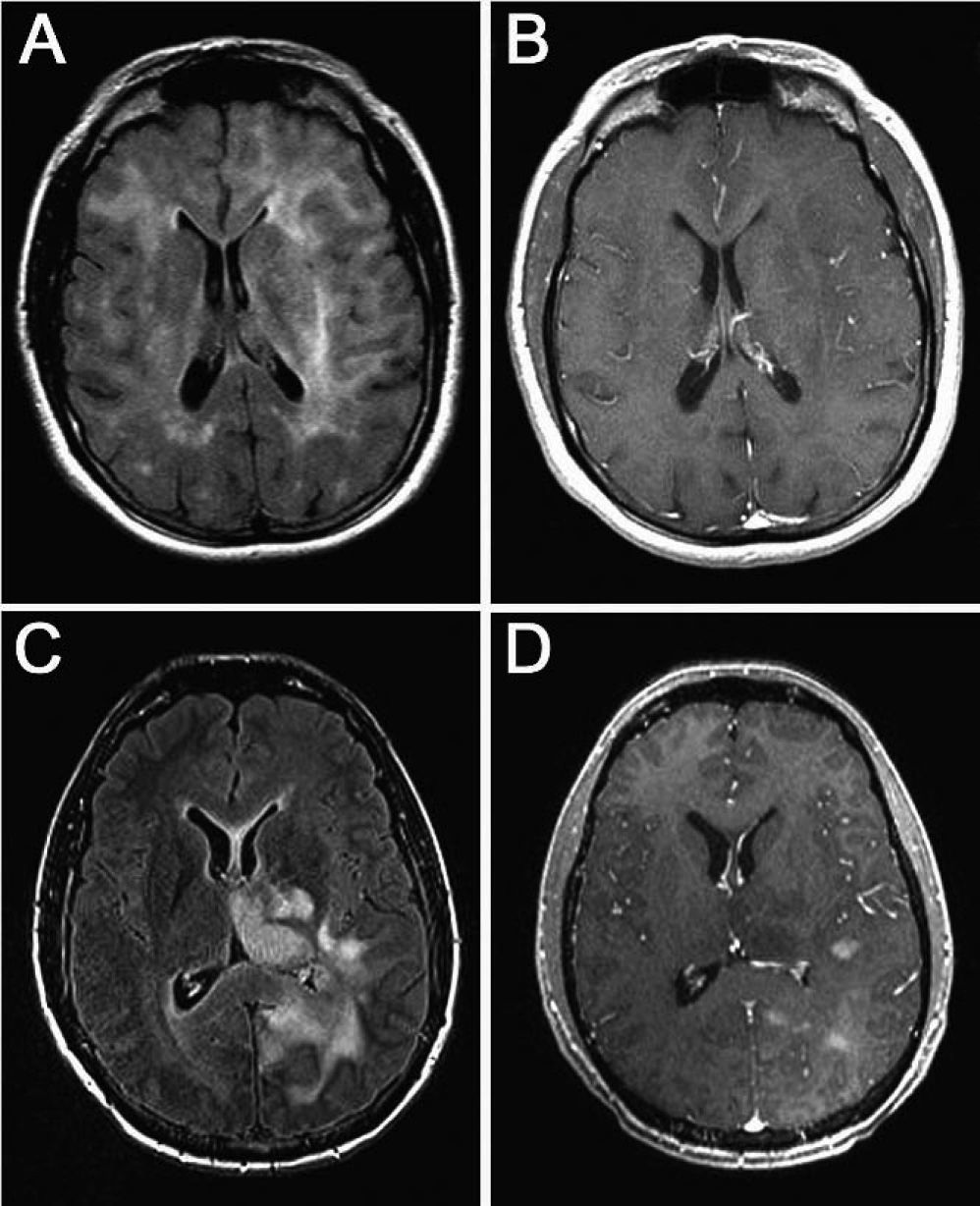
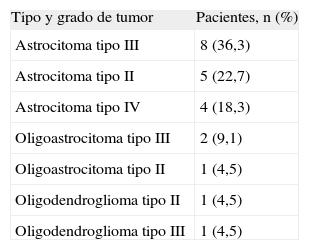
ResultadosEn total, identificamos a 22 pacientes diagnosticados de GC, 17 varones (77,2%) y 5 mujeres (22,8%) (tabla 1). La media de edad fue 54 años (51 en varones y 54 en mujeres; intervalo, 23-81 años). Las manifestaciones clínicas iniciales fueron déficit focales sensitivo-motores o visuales (86,4%), crisis epilépticas (36,4%), deterioro cognitivo subagudo (27,3%) y cefalea (27,3%). Todos los pacientes tenían una afección radiológica bilateral (figs. 1 y 2), con deterioro de más de 2 lóbulos cerebrales, que involucraba temporal (86,4%), frontal (77,2%), parietal (77,2%) y occipital (40,9%); otras estructuras afectadas fueron los ganglios basales (81,8%), cuerpo calloso (45,4%), troncoencéfalo (22,7%), ínsula (13,6%), cerebelo (9,1%), médula cervical (4,5%), pares craneales (4,5%) y leptomeninges (4,5%). Todos los casos correspondieron a GC primarias: 16 pacientes presentaban el tipo 1 y 6, el tipo 2, la mayoría de estos últimos con efecto de masa concomitante. La anatomía patológica demostró astrocitoma de grado III en 8 pacientes (36,4%), astrocitoma de grado II en 5 (22,7%), astrocitoma con características de grado IV en 4 (18,3%), oligoastrocitoma de grado III en 2 (9,1%), oligoastrocitoma de grado II en 1 (4,5%), oligodendroglioma de grado II en 1 (4,5%) y oligodendroglioma de grado III en 1 (4,5%) (tabla 2). El tiempo medio de retraso diagnóstico fue 1 mes; 9 pacientes (40,9%) fueron diagnosticados durante el primer mes tras iniciarse los síntomas; 6 (27,3%), durante el segundo mes; 5 (22,7%), entre el segundo mes y el primer año, y 2 (9,1%), después de 1 año del inicio de los síntomas. En estos 2 pacientes los síntomas se atribuyeron a migraña en un caso y en el otro a encefalitis límbica de origen desconocido5. En 4 pacientes el diagnóstico inicial considerado fue de un proceso cerebrovascular agudo.
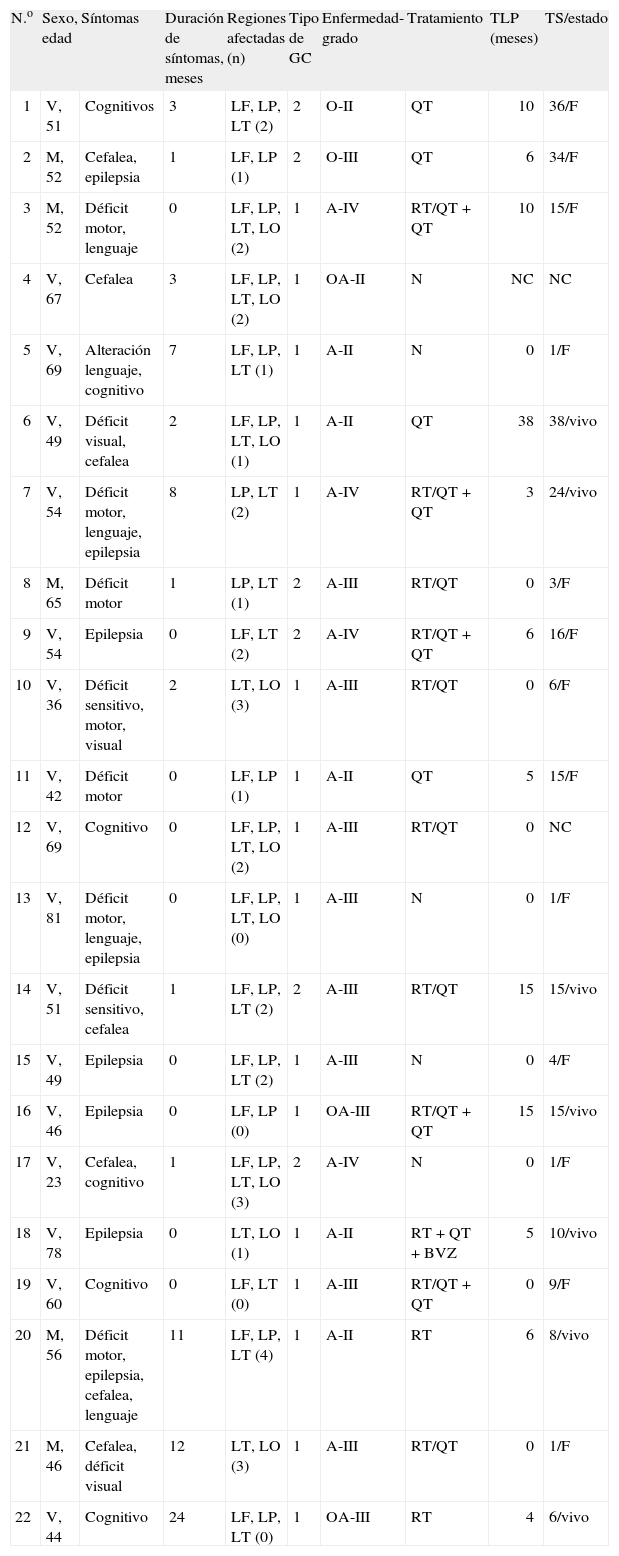
Características clínicas, tratamiento y evolución
| N.o | Sexo, edad | Síntomas | Duración de síntomas, meses | Regiones afectadas (n) | Tipo de GC | Enfermedad-grado | Tratamiento | TLP (meses) | TS/estado |
| 1 | V, 51 | Cognitivos | 3 | LF, LP, LT (2) | 2 | O-II | QT | 10 | 36/F |
| 2 | M, 52 | Cefalea, epilepsia | 1 | LF, LP (1) | 2 | O-III | QT | 6 | 34/F |
| 3 | M, 52 | Déficit motor, lenguaje | 0 | LF, LP, LT, LO (2) | 1 | A-IV | RT/QT + QT | 10 | 15/F |
| 4 | V, 67 | Cefalea | 3 | LF, LP, LT, LO (2) | 1 | OA-II | N | NC | NC |
| 5 | V, 69 | Alteración lenguaje, cognitivo | 7 | LF, LP, LT (1) | 1 | A-II | N | 0 | 1/F |
| 6 | V, 49 | Déficit visual, cefalea | 2 | LF, LP, LT, LO (1) | 1 | A-II | QT | 38 | 38/vivo |
| 7 | V, 54 | Déficit motor, lenguaje, epilepsia | 8 | LP, LT (2) | 1 | A-IV | RT/QT + QT | 3 | 24/vivo |
| 8 | M, 65 | Déficit motor | 1 | LP, LT (1) | 2 | A-III | RT/QT | 0 | 3/F |
| 9 | V, 54 | Epilepsia | 0 | LF, LT (2) | 2 | A-IV | RT/QT + QT | 6 | 16/F |
| 10 | V, 36 | Déficit sensitivo, motor, visual | 2 | LT, LO (3) | 1 | A-III | RT/QT | 0 | 6/F |
| 11 | V, 42 | Déficit motor | 0 | LF, LP (1) | 1 | A-II | QT | 5 | 15/F |
| 12 | V, 69 | Cognitivo | 0 | LF, LP, LT, LO (2) | 1 | A-III | RT/QT | 0 | NC |
| 13 | V, 81 | Déficit motor, lenguaje, epilepsia | 0 | LF, LP, LT, LO (0) | 1 | A-III | N | 0 | 1/F |
| 14 | V, 51 | Déficit sensitivo, cefalea | 1 | LF, LP, LT (2) | 2 | A-III | RT/QT | 15 | 15/vivo |
| 15 | V, 49 | Epilepsia | 0 | LF, LP, LT (2) | 1 | A-III | N | 0 | 4/F |
| 16 | V, 46 | Epilepsia | 0 | LF, LP (0) | 1 | OA-III | RT/QT + QT | 15 | 15/vivo |
| 17 | V, 23 | Cefalea, cognitivo | 1 | LF, LP, LT, LO (3) | 2 | A-IV | N | 0 | 1/F |
| 18 | V, 78 | Epilepsia | 0 | LT, LO (1) | 1 | A-II | RT + QT + BVZ | 5 | 10/vivo |
| 19 | V, 60 | Cognitivo | 0 | LF, LT (0) | 1 | A-III | RT/QT + QT | 0 | 9/F |
| 20 | M, 56 | Déficit motor, epilepsia, cefalea, lenguaje | 11 | LF, LP, LT (4) | 1 | A-II | RT | 6 | 8/vivo |
| 21 | M, 46 | Cefalea, déficit visual | 12 | LT, LO (3) | 1 | A-III | RT/QT | 0 | 1/F |
| 22 | V, 44 | Cognitivo | 24 | LF, LP, LT (0) | 1 | OA-III | RT | 4 | 6/vivo |
A: astrocitoma; BVZ: bevacizumab; F: fallecido; GC: gliomatosis cerebral; LF: lóbulo frontal; LO: lóbulo occipital; LP: lóbulo parietal; LT: lóbulo temporal; M: mujer; N: sin tratamiento; NC: no conocido; O: oligodendroglioma; OA: oligoastrocitoma; QT: quimioterapia; RT: radioterapia; TLP: tiempo libre de progresión; TS: tiempo de supervivencia; V: varón.

 en la que observamos diferente grado de captación de las lesiones. A y B: imágenes de un mismo paciente en el que la infiltración no capta contraste a pesar de la extensa afección. C y D: imágenes de otro paciente en el que la lesión capta contraste de manera difusa.")
Resonancia magnética con secuencias FLAIR y T1 tras administración de contraste (gadolinio intravenoso) en la que observamos diferente grado de captación de las lesiones. A y B: imágenes de un mismo paciente en el que la infiltración no capta contraste a pesar de la extensa afección. C y D: imágenes de otro paciente en el que la lesión capta contraste de manera difusa.
Diecisiete pacientes tuvieron un seguimiento periódico, 4 fallecieron poco después del diagnóstico y 1 paciente no volvió a las consultas. Los tratamientos administrados fueron quimiorradioterapia (RT/QT) con 75mg/m2/día de temozolamida (TMZ) oral y radioterapia (RT) estándar fraccionada seguida de ciclos de TMZ (150-200mg/m2/día durante 5 días, cada 28 días) en 5 pacientes, RT/QT en otros 5, únicamente TMZ en 4, RT sola en 2 y RT seguida de TMZ más bevacizumab (BVZ) en 1. Se trató a 5 de los 7 pacientes con tumores de bajo grado y todos respondieron al tratamiento. Nueve de los 11 pacientes con tumores de grado III recibieron tratamiento: 4 (44,4%) respondieron y 5 no respondieron. Tres de los 4 pacientes con áreas de grado IV recibieron tratamiento y todos respondieron. En total, el 70,6% de los pacientes tratados respondieron al tratamiento. Nueve pacientes (75%) tuvieron progresión tumoral, con un tiempo libre de progresión de 6 meses; 8 recibieron tratamiento (4, TMZ a dosis diarias de 75mg/ m2; 2, RT/QT; 1, procarbazina/lomustina/vincristina, y 1, BVZ/irinotecán; 2 pacientes necesitaron cirugía resectiva del área de progresión). De estos 8 pacientes, 7 (87,5%) tuvieron respuesta terapéutica. Seis pacientes (85,7%) evidenciaron una segunda progresión y fueron tratados con TMZ (2), BVZ (1), irinotecán (1), BVZ/irinotecán (1) o RT más etopósido/erlotinib (1) y 3 de ellos (50%) respondieron al tratamiento.
De los 22 pacientes, 13 han fallecido, 7 siguen vivos y de 2 no hay seguimiento. La media de seguimiento fue 13 meses. Ocho pacientes (36,3%) desarrollaron complicaciones tromboembólicas (6, trombosis venosa profunda; 5, embolia pulmonar, y 2, trombosis venosa cerebral); un mismo paciente desarrolló un hematoma epidural y subdural. La supervivencia general fue de 9,5 meses, que aumentó a 15 meses en los pacientes tratados.
DiscusiónLa serie más larga de GC fue publicada por Taillibert et al2 en 2006 e incluye a 296 pacientes. La mayoría eran varones (56,8%) y la media de edad de los pacientes fue 39 años (39 años en varones y 45 en mujeres), con casos aislados de recién nacidos y ancianos. En nuestra serie, la proporción de varones también es mayor (77,2%) y la media de edad es 54 años (51 años en varones y 54 en mujeres); debido a que es un centro de adultos no hay pacientes pediátricos. En la serie de Taillibert et al2, el 31,1% de los pacientes comenzaron con crisis epilépticas (el 36,4% en nuestra serie), un 18,6% con deterioro cognitivo (el 27,3% en nuestra serie) y el 16,9% con déficit focales (el 86,4% en nuestra serie). La diferencia en los déficit focales puede deberse a que en el estudio de Taillibert no están recogidos los síntomas iniciales de todos los pacientes y que en algunos de nuestros pacientes la historia clínica menciona varios síntomas como el primero, sin diferenciar cuál fue el realmente inicial. En nuestra serie la mayoría de los pacientes tenían GC primaria tipo 1 (72,7%), lo cual contrasta con una serie de 33 pacientes de Park et al6, en que la GC tipo 2 era más frecuente (54,5%). En nuestra serie, la GC tipo 2 puede estar subestimada por la dificultad en ocasiones de diferenciar la masa tumoral per se de la infiltración. La afectación radiológica fue de predominio hemisférico, aunque destaca la afección diencefálica en el 81,8% de los pacientes, semejante a otros estudios como el de Vates et al7, con afección diencefálica en 21 de 22 pacientes (95%). En el estudio de Taillibert et al2, el 60,3% de los tumores eran astrocitomas (el 67,2% en el nuestro), un 50% eran tumores de grado II (el 31,7% en nuestra serie), el 40,5%, de grado III (el 49,9% en nuestra serie) y un 9,5% mostró áreas de grado IV (el 18,3% en nuestra serie), lo que indica una mayor agresividad histológica en nuestros pacientes. Aunque en 2 pacientes los síntomas se atribuyeron a otros procesos neurológicos, lo cual retrasó el diagnóstico en más de 1 año, en general la media de tiempo en realizar el diagnóstico fue 1 mes, considerablemente inferior a los 3 meses del estudio de Vates et al7. El diagnóstico diferencial de la GC suele incluir las encefalitis infecciosas o autoinmunitarias, enfermedades desmielinizantes, vasculitis y otros tumores cerebrales, como el linfoma cerebral primario2,3,7. En nuestros pacientes el diagnóstico inicial más frecuentemente considerado fue un proceso cerebrovascular agudo.
El tratamiento de la GC no está bien establecido, por lo que los tratamientos administrados a los pacientes no siguieron ningún protocolo concreto. La cirugía es diagnóstica y paliativa. La RT mejora los síntomas neurológicos, pero está limitada por su toxicidad al aplicarla sobre un volumen cerebral extenso, y su efecto en la supervivencia difiere según los estudios8–10. La QT, fundamentalmente TMZ, ha mostrado respuesta clínica, radiológica y mejora de la supervivencia11–13.
No hemos encontrado en la literatura estudios que evalúen el efecto de la RT/QT, con o sin QT adyuvante, en la GC. El 70,6% de los pacientes respondieron inicialmente al tratamiento, aunque el 75% progresó posteriormente en una media de 6 meses. El 87,5% respondió al tratamiento de la primera recidiva tumoral y un 50% respondió al tratamiento de una segunda recidiva. La supervivencia de los pacientes tratados fue de 15 meses, cifra que supera en 5,5 meses la supervivencia general de la muestra (9,5 meses) y la de los pacientes con glioblastoma tratados de manera estándar con RT/QT y QT adyuvante según el protocolo de Stupp et al14 (14,5 meses). Destaca 1 paciente con hemianopsia estable y astrocitoma de bajo grado histológico que recibió TMZ y se mantiene sin progresión 38 meses después del diagnóstico. Las complicaciones más frecuentes fueron hemáticas, sobre todo tromboembólicas, posiblemente debido a la activación sistémica de la coagulación por el propio tumor.
Nuestro estudio y los de otros investigadores indican que los pacientes con GC frecuentemente responden al tratamiento2,7–13, incluso después de 1 o 2 recidivas, con una supervivencia similar a la de los pacientes con glioblastomas14.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

