



Hoy en día se acepta que el sistema nervioso central adulto posee una enorme flexibilidad morfofuncional que le permite realizar procesos de remodelación estructural aún después de haber alcanzado su desarrollo y maduración. Además del enorme número de genes que participan en el desarrollo de la memoria, los diferentes mecanismos epigenéticos conocidos también han sido involucrados en procesos de modificación neuronal normal y patológica y, por ende, en los mecanismos de desarrollo de la memoria.
DesarrolloEste trabajo fue llevado a cabo a través de una sistemática revisión de las bases de datos de publicaciones biomédicas sobre los aspectos genéticos y epigenéticos que participan en la función sináptica y la memoria.
ConclusionesLa activación de la expresión génica, en respuesta a estímulos extrínsecos, ocurre también en células nerviosas diferenciadas. La actividad neuronal induce formas específicas de plasticidad sináptica que permiten la formación y almacenamiento de la memoria a largo plazo. Los mecanismos epigenéticos tienen un papel crucial en los procesos de modificación sináptica y en la formación y desarrollo de la memoria. Alteraciones en estos mecanismos producen déficit cognitivo y de memoria en padecimientos neurodegenerativos (enfermedad de Alzheimer y Huntington) así como en trastornos del desarrollo neurológico (síndrome de Rett, X-frágil y esquizofrenia). Los resultados obtenidos en diferentes modelos muestran, sin embargo, un escenario promisorio con tratamientos potenciales para algunos de estos padecimientos.
Today, scientists accept that the central nervous system of an adult possesses considerable morphological and functional flexibility, allowing it to perform structural remodelling processes even after the individual is fully developed and mature. In addition to the vast number of genes participating in the development of memory, different known epigenetic mechanisms are involved in normal and pathological modifications to neurons and therefore also affect the mechanisms of memory development.
DevelopmentThis study entailed a systematic review of biomedical article databases in search of genetic and epigenetic factors that participate in synaptic function and memory.
ConclusionsThe activation of gene expression in response to external stimuli also occurs in differentiated nerve cells. Neural activity induces specific forms of synaptic plasticity that permit the creation and storage of long-term memory. Epigenetic mechanisms play a key role in synaptic modification processes and in the creation and development of memory. Changes in these mechanisms result in the cognitive and memory impairment seen in neurodegenerative diseases (Alzheimer disease, Huntington disease) and in neurodevelopmental disorders (Rett syndrome, fragile X, and schizophrenia). Nevertheless, results obtained from different models are promising and point to potential treatments for some of these diseases.
Por mucho tiempo se consideró que el cerebro de mamíferos adultos era un órgano incapaz de continuar con procesos de remodelación estructural luego de terminadas sus etapas del desarrollo. Tal concepto se aplicaba a todas las estructuras del sistema nervioso y en especial a las sinapsis. En los últimos años estos conceptos han cambiado drásticamente, al punto de aceptar que existe una enorme flexibilidad en la forma y función neuronal del cerebro adulto. Las neuronas y sinapsis sufren diversas formas de plasticidad estructural y funcional, permitiendo cambios profundos en la estructura cerebral íntima. Estos cambios son adaptables y generados principalmente por los patrones de actividad neuronal, que son a su vez estimulados por la experiencia sensorial tanto de fuentes internas como externas1.
Los recientes descubrimientos de la biología del desarrollo mostraron que la activación de la expresión génica en respuesta a señales extracelulares, del tipo de los factores de crecimiento, ocurre también en células posmitóticas diferenciadas. La activación transcripcional en células maduras puede ser inducida por una variedad de estímulos extrínsecos, incluyendo algunos de los factores que activan la transcripción durante el desarrollo embrionario y fetal. En particular se ha demostrado que las neuronas pueden modificar la expresión de un grupo de genes en respuesta a estímulos de despolarización, lo cual permitió plantear la hipótesis que la actividad génica puede ser influenciada por la actividad sináptica normal.
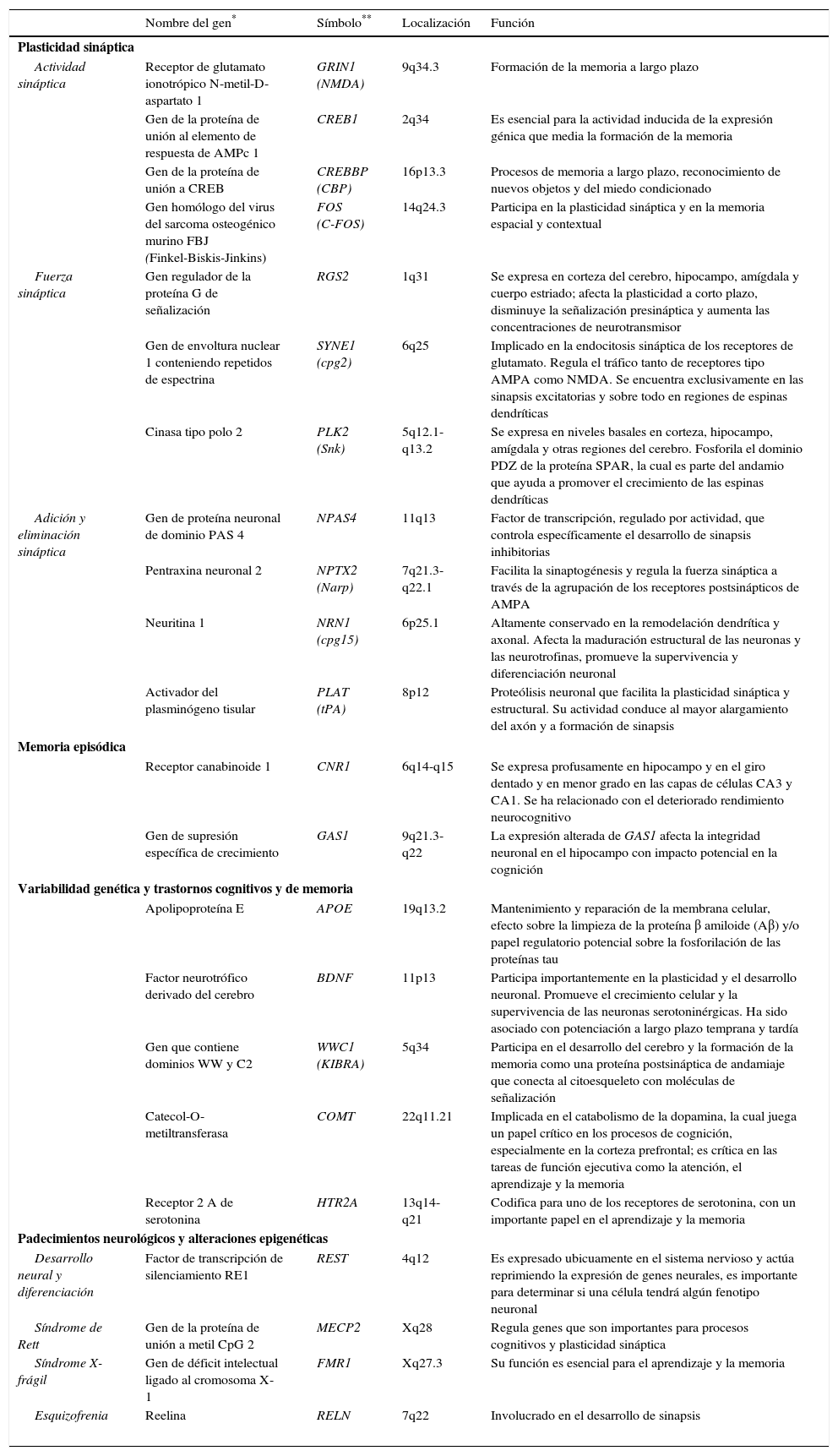
La observación de que la actividad neuronal induce tanto los cambios adaptativos neuronales como los cambios en los patrones de expresión génica, sugieren que tal secuencia de eventos es el origen de formas específicas de plasticidad neuronal y específicamente sináptica. También se ha logrado demostrar que los mismos genes que son estimulados por la actividad sináptica funcionan también durante el desarrollo del cerebro, lo que sugiere que la plasticidad, en todas las etapas del desarrollo cerebral, utiliza mecanismos y maquinaria molecular similar2 (tabla 1).
Genes implicados en plasticidad sináptica, aprendizaje y memoria
| Nombre del gen* | Símbolo** | Localización | Función | |
|---|---|---|---|---|
| Plasticidad sináptica | ||||
| Actividad sináptica | Receptor de glutamato ionotrópico N-metil-D-aspartato 1 | GRIN1 (NMDA) | 9q34.3 | Formación de la memoria a largo plazo |
| Gen de la proteína de unión al elemento de respuesta de AMPc 1 | CREB1 | 2q34 | Es esencial para la actividad inducida de la expresión génica que media la formación de la memoria | |
| Gen de la proteína de unión a CREB | CREBBP (CBP) | 16p13.3 | Procesos de memoria a largo plazo, reconocimiento de nuevos objetos y del miedo condicionado | |
| Gen homólogo del virus del sarcoma osteogénico murino FBJ (Finkel-Biskis-Jinkins) | FOS (C-FOS) | 14q24.3 | Participa en la plasticidad sináptica y en la memoria espacial y contextual | |
| Fuerza sináptica | Gen regulador de la proteína G de señalización | RGS2 | 1q31 | Se expresa en corteza del cerebro, hipocampo, amígdala y cuerpo estriado; afecta la plasticidad a corto plazo, disminuye la señalización presináptica y aumenta las concentraciones de neurotransmisor |
| Gen de envoltura nuclear 1 conteniendo repetidos de espectrina | SYNE1 (cpg2) | 6q25 | Implicado en la endocitosis sináptica de los receptores de glutamato. Regula el tráfico tanto de receptores tipo AMPA como NMDA. Se encuentra exclusivamente en las sinapsis excitatorias y sobre todo en regiones de espinas dendríticas | |
| Cinasa tipo polo 2 | PLK2 (Snk) | 5q12.1-q13.2 | Se expresa en niveles basales en corteza, hipocampo, amígdala y otras regiones del cerebro. Fosforila el dominio PDZ de la proteína SPAR, la cual es parte del andamio que ayuda a promover el crecimiento de las espinas dendríticas | |
| Adición y eliminación sináptica | Gen de proteína neuronal de dominio PAS 4 | NPAS4 | 11q13 | Factor de transcripción, regulado por actividad, que controla específicamente el desarrollo de sinapsis inhibitorias |
| Pentraxina neuronal 2 | NPTX2 (Narp) | 7q21.3-q22.1 | Facilita la sinaptogénesis y regula la fuerza sináptica a través de la agrupación de los receptores postsinápticos de AMPA | |
| Neuritina 1 | NRN1 (cpg15) | 6p25.1 | Altamente conservado en la remodelación dendrítica y axonal. Afecta la maduración estructural de las neuronas y las neurotrofinas, promueve la supervivencia y diferenciación neuronal | |
| Activador del plasminógeno tisular | PLAT (tPA) | 8p12 | Proteólisis neuronal que facilita la plasticidad sináptica y estructural. Su actividad conduce al mayor alargamiento del axón y a formación de sinapsis | |
| Memoria episódica | ||||
| Receptor canabinoide 1 | CNR1 | 6q14-q15 | Se expresa profusamente en hipocampo y en el giro dentado y en menor grado en las capas de células CA3 y CA1. Se ha relacionado con el deteriorado rendimiento neurocognitivo | |
| Gen de supresión específica de crecimiento | GAS1 | 9q21.3-q22 | La expresión alterada de GAS1 afecta la integridad neuronal en el hipocampo con impacto potencial en la cognición | |
| Variabilidad genética y trastornos cognitivos y de memoria | ||||
| Apolipoproteína E | APOE | 19q13.2 | Mantenimiento y reparación de la membrana celular, efecto sobre la limpieza de la proteína β amiloide (Aβ) y/o papel regulatorio potencial sobre la fosforilación de las proteínas tau | |
| Factor neurotrófico derivado del cerebro | BDNF | 11p13 | Participa importantemente en la plasticidad y el desarrollo neuronal. Promueve el crecimiento celular y la supervivencia de las neuronas serotoninérgicas. Ha sido asociado con potenciación a largo plazo temprana y tardía | |
| Gen que contiene dominios WW y C2 | WWC1 (KIBRA) | 5q34 | Participa en el desarrollo del cerebro y la formación de la memoria como una proteína postsináptica de andamiaje que conecta al citoesqueleto con moléculas de señalización | |
| Catecol-O-metiltransferasa | COMT | 22q11.21 | Implicada en el catabolismo de la dopamina, la cual juega un papel crítico en los procesos de cognición, especialmente en la corteza prefrontal; es crítica en las tareas de función ejecutiva como la atención, el aprendizaje y la memoria | |
| Receptor 2 A de serotonina | HTR2A | 13q14-q21 | Codifica para uno de los receptores de serotonina, con un importante papel en el aprendizaje y la memoria | |
| Padecimientos neurológicos y alteraciones epigenéticas | ||||
| Desarrollo neural y diferenciación | Factor de transcripción de silenciamiento RE1 | REST | 4q12 | Es expresado ubicuamente en el sistema nervioso y actúa reprimiendo la expresión de genes neurales, es importante para determinar si una célula tendrá algún fenotipo neuronal |
| Síndrome de Rett | Gen de la proteína de unión a metil CpG 2 | MECP2 | Xq28 | Regula genes que son importantes para procesos cognitivos y plasticidad sináptica |
| Síndrome X-frágil | Gen de déficit intelectual ligado al cromosoma X-1 | FMR1 | Xq27.3 | Su función es esencial para el aprendizaje y la memoria |
| Esquizofrenia | Reelina | RELN | 7q22 | Involucrado en el desarrollo de sinapsis |
Más recientemente se demostró que la expresión génica, subyacente a la actividad sináptica y neuronal, se encuentra regulada en gran medida por mecanismos epigenéticos de la misma índole que los que ocurren en procesos del desarrollo embrionario o fetal. Las evidencias sugieren que las modificaciones epigenéticas dentro del SNC, que son cruciales para la adaptación de la conducta a corto y largo plazo, ocurren como consecuencia de diversos estímulos ambientales. La activación o silenciamiento de estos genes, controlados por mecanismos epigenéticos, parece representar un importante regulador del potencial sináptico y la memoria.
Este trabajo tiene como objetivo presentar una revisión sistemática y actualizada de los mecanismos epigenéticos relacionados con la función sináptica y con la memoria, así como de las alteraciones epigenéticas involucradas en la afectación de la memoria, tanto en alteraciones del neurodesarrollo como en padecimientos neurodegenerativos.
Mecanismos epigenéticosLa epigenética es un término acuñado por Waddington para referirse al conjunto de procesos de regulación de la expresión génica, que no incurren en cambios en la secuencia de nucleótidos del ADN y que tienen un carácter heredable3. Hasta la fecha se han descrito 3 mecanismos que participan en forma importante en la regulación génica: 1) la modificación de histonas, 2) la metilación del ADN y, 3) los ácidos ribonucleicos (ARN) no codificantes. Entre estos, la modificación de histonas es el proceso más ampliamente conocido y relacionado con un importante número de enfermedades neurológicas4–6.
Modificación de histonasLas histonas son proteínas básicas que participan en el empaquetamiento del ADN y formación de los nucleosomas. La modificación de las histonas es un mecanismo que ocurre en forma independiente o como consecuencia de la metilación del ADN6,7. La hipótesis actual indica que los extremos amino terminales de las histonas, que sobresalen de la estructura de la cromatina, participan en un proceso de integración de señales que dan lugar a un patrón específico de modificaciones postraduccionales y forman un «código de histonas», el cual dirige la actividad de numerosos factores y cofactores de la maquinaria transcripcional. Actualmente se conocen 4 tipos de modificaciones postraduccionales en los extremos de las proteínas histónicas que participan en el marcaje epigenético: 1) acetilación, 2) metilación, 3) ubiquitinación y 4) fosforilación6,8,9.
Metilación del ADNEl ADN presenta regiones de 1.000-1.500 pares de bases ricas en dinucleotidos CpG (islas CpG), que son reconocidas por las enzimas ADN-metiltransferasas. Durante la replicación del ADN, las citosinas de la cadena recién sintetizada son metiladas, manteniéndose así la memoria del estado metilado en la molécula hija de ADN. En general esta modificación se hace estable y es heredada como un patrón clonal de metilación. La sobremetilación de estas regiones mantiene la estructura condensada de la cromatina e impide la transcripción de los genes involucrados6,8,9.
ARN no codificantesNo codifican para ninguna proteína pero sus secuencias son complementarias a ADN o ARN codificante e impiden su traducción; esta es una forma de regulación negativa de la expresión a nivel postranscripcional. A este tipo pertenecen los denominados ARN de interferencia (iARN) los cuales se unen a secuencias complementarias de ARNm para degradarlo e impedir su traducción en proteínas6,8,9.
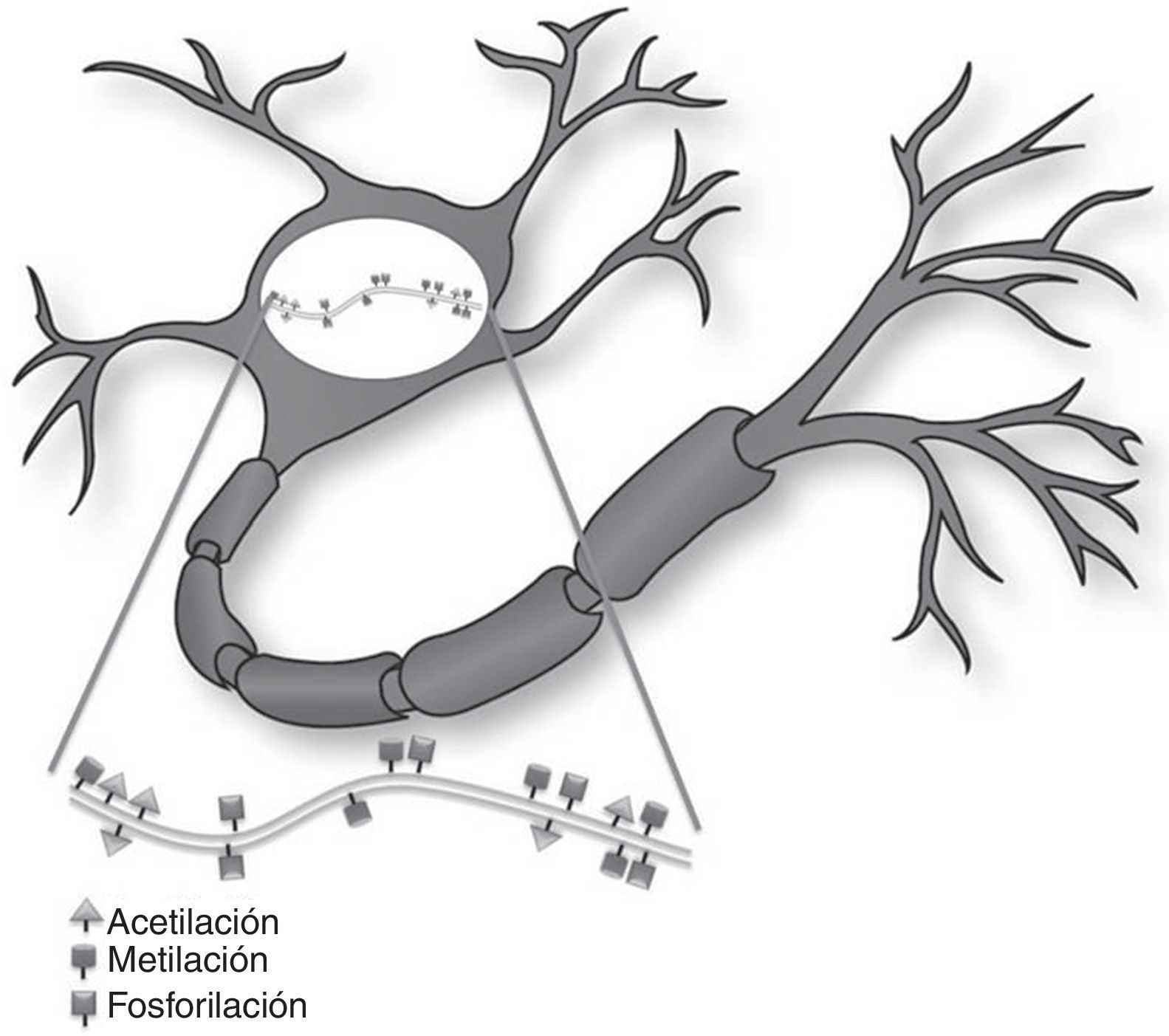
Mecanismos epigenéticos en la función sináptica y formación de la memoriaLa memoria se describe como el proceso cerebral que permite almacenar información a largo plazo8. Recientes evidencias sugieren que una amplia variedad de estímulos ambientales producen modificaciones epigenéticas en el SNC que son críticas para la adaptación de la conducta a corto y largo plazo (fig. 1)10. Como tal, las modificaciones epigenéticas intervienen por diferentes mecanismos en la creación y mantenimiento de la memoria conductual en múltiples niveles. Estos y otros estudios han generado interés en el potencial terapéutico de fármacos capaces de mejorar o alterar estas reacciones en los trastornos cognitivos8,11.

Control epigenético en el sistema nervioso del adulto. La regulación epigenética en neuronas de cerebro adulto ocurre en respuesta a señales sinápticas y/o estímulos ambientales. Estos estímulos externos resultan en cambios en el perfil transcripcional y por ende en la función neuronal.
El estudio llevado a cabo por Levenson et al. demostró que la formación de la memoria a largo plazo también está sujeta a marcaje epigenético. En el modelo de acondicionamiento al miedo contextual un modelo de aprendizaje dependiente del hipocampo en el que el animal aprende a asociarse a un nuevo ambiente con un estímulo aversivo, la acetilación de la histona H3, pero no de H4, se observa significativamente incrementada12. La formación de la memoria a largo plazo en el modelo del miedo contextual requiere del receptor de glutamato ionotrópico N-metil-D-aspartato 1(GRIN1) y de la cascada de señalización MEK-ERK/MAPK en el hipocampo; la inhibición de cualquiera de estos, bloquea la acetilación de la histona H3. Estos hallazgos indican que, en el código de histonas, tipos específicos de memoria se asocian con patrones específicos de modificaciones histónicas12.
Otros autores han investigado la formación de la memoria a largo plazo en ratones transgénicos con alteraciones en la función de las proteínas de unión a CREB (CBP). Estos ratones transgénicos son heterocigotos para una forma dominante negativa o truncada de CBP (CBPDN+/-)105, y tienen un déficit significativo en varias formas de memoria a largo plazo, como la de evitar el cruzamiento pasivo de espacios, el reconocimiento de nuevos objetos y el de miedo condicionado13.
Otros 2 estudios independientes utilizaron ratones deficientes en CBP, pero que carecían de los graves problemas del desarrollo de los ratones CBPDN+/-. En el primero de estos estudios se transformó el alelo dominante negativo de CBP a un promotor inducible (CBPI-DN+/−)14. La activación del alelo dominante negativo, posterior al desarrollo normal de los animales, mostró alteración en el aprendizaje de los laberintos espaciales de agua y en el reconocimiento de nuevos objetos14. En el segundo estudio, ratones heterocigotos para CBP (CBP+/−) tuvieron alteraciones en la memoria contextual y en la memoria condicionada al miedo y mostraron alteraciones en el reconocimiento de nuevos objetos15. En ambos estudios, la administración de un inhibidor de la enzima desacetilasa de histona (HDAC) restauró la formación de la memoria a largo plazo. Esto significa que cualquier alteración en los procesos que regulan la estructura de la cromatina puede afectar la formación de la memoria a largo plazo in vivo12. En ambos estudios, los inhibidores de la HDAC no afectaron la memoria a corto plazo8.
Plasticidad sinápticaLos cambios en la fuerza de la actividad sináptica son ampliamente aceptados como críticos en la formación de la memoria a largo plazo. Muchos estudios han caracterizado los mecanismos que son responsables para la inducción, expresión y mantenimiento de la plasticidad sináptica en varios organismos16. Una interesante observación es que estos mecanismos son similares a aquellos involucrados en la formación de la memoria a largo plazo. Así, la inducción de la plasticidad sináptica quizás involucre mecanismos epigenéticos similares a los involucrados en la memoria a largo plazo. Un ejemplo de este mecanismo de sinapsis sensorial y motora se describió en el molusco marino Aplysia que muestra 2 formas de plasticidad: la facilitación a largo plazo (FLP) que se refiere al mejoramiento duradero de la transmisión sináptica y la depresión a largo plazo (DLP) que es una disminución duradera en la transmisión sináptica. La acetilación de la histona H4 en el promotor de la proteína de unión CCAAT (C/EBP) de Aplysia fue transitoriamente incrementada durante la FLP, pero también disminuyó transitoriamente durante la DLP. Por lo tanto, estas 2 formas opuestas de plasticidad inducen cambios opuestos en la acetilación de las histonas en Aplysia17.
Los cambios epigenéticos inducidos por la plasticidad sináptica son observados también en modelos mamíferos. La activación directa de los receptores GRIN1 en el hipocampo incrementa la acetilación de las histonas H3, las cuales pueden ser bloqueadas por la inhibición de la cascada MEK-ERK/MAPK. De igual forma, la activación de vías de señalización dopaminérgicas, colinérgicas y glutamatérgicas en el hipocampo induce la fosforilación de las histonas H3 dependientes del incremento de ERK12. Estos resultados sugieren que la inducción de la plasticidad sináptica en los mamíferos genera un incremento en la acetilación y fosforilación de las histonas en el hipocampo dependiente de ERK, de manera similar a lo observado en Aplysia. Estos estudios indican que el estado epigenético del genoma afecta la inducción a largo plazo en la plasticidad sináptica en los mamíferos8,12.
Enfermedades neurológicas y alteraciones epigenéticasExiste un conjunto de evidencias que implican a los mecanismos epigenéticos como causa de disfunciones cognitivas humanas. Padecimientos neurodegenerativos y enfermedades del neurodesarrollo pueden ser atribuidos, al menos en parte, a mecanismos subyacentes al marcaje epigenético del genoma. Se analizarán las evidencias encontradas en algunos de los padecimientos más interesantes.
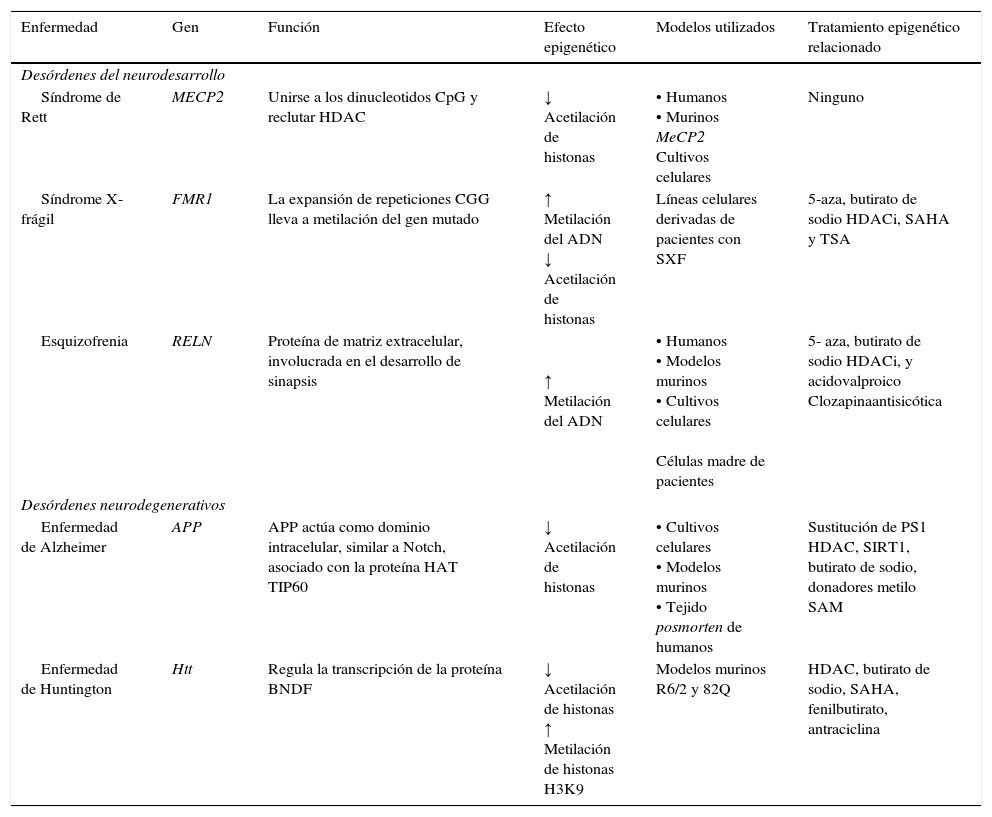
Padecimientos neurodegenerativosAlgunos padecimientos neurodegenerativos parecen estar relacionados con alteraciones en los mecanismos epigenéticos; entre ellos, la enfermedad de Alzheimer (EA) y la enfermedad de Huntington (EH) son intensamente estudiados (tabla 2).
Mecanismos y terapias epigenéticas en padecimientos neurodegenerativos y del neurodesarrollo
| Enfermedad | Gen | Función | Efecto epigenético | Modelos utilizados | Tratamiento epigenético relacionado |
|---|---|---|---|---|---|
| Desórdenes del neurodesarrollo | |||||
| Síndrome de Rett | MECP2 | Unirse a los dinucleotidos CpG y reclutar HDAC | ↓ Acetilación de histonas | • Humanos • Murinos MeCP2 Cultivos celulares | Ninguno |
| Síndrome X- frágil | FMR1 | La expansión de repeticiones CGG lleva a metilación del gen mutado | ↑ Metilación del ADN ↓ Acetilación de histonas | Líneas celulares derivadas de pacientes con SXF | 5-aza, butirato de sodio HDACi, SAHA y TSA |
| Esquizofrenia | RELN | Proteína de matriz extracelular, involucrada en el desarrollo de sinapsis | ↑ Metilación del ADN | • Humanos • Modelos murinos • Cultivos celulares Células madre de pacientes | 5- aza, butirato de sodio HDACi, y acidovalproico Clozapinaantisicótica |
| Desórdenes neurodegenerativos | |||||
| Enfermedad de Alzheimer | APP | APP actúa como dominio intracelular, similar a Notch, asociado con la proteína HAT TIP60 | ↓ Acetilación de histonas | • Cultivos celulares • Modelos murinos • Tejido posmorten de humanos | Sustitución de PS1 HDAC, SIRT1, butirato de sodio, donadores metilo SAM |
| Enfermedad de Huntington | Htt | Regula la transcripción de la proteína BNDF | ↓ Acetilación de histonas ↑ Metilación de histonas H3K9 | Modelos murinos R6/2 y 82Q | HDAC, butirato de sodio, SAHA, fenilbutirato, antraciclina |
APP: proteína precursora amiloide; CREBBP: proteína de unión a CREB1; EP300: proteína de unión p300; HDAC: histona desacetilasa; HDACi: inhibidor de histonas desacetilasas; HAT: histona acetiltransferasa; Htt: hungtintina; MECP2: proteína de unión a metilCpG; PS1: presenilina; SAHA: acidosuberoilsanilidohidroxalamico; SIRT1: regulador homologo 1 de información silente; TSA: tricostatina A; 5-aza: 5- aza-2-deoxicitidina. Modificado de: Levenson y Sweatt8, Scarpa et al.24
Es la causa más frecuente de demencia degenerativa primaria. Se caracteriza por alteraciones cognitivas, demencia y por la acumulación de placas formadas por fragmentos β-amiloides y agregados neurofibrilares en diferentes regiones del cerebro. Su prevalencia es de alrededor de 35 millones de personas en el mundo18,19.
Recientes evidencias demuestran que la acetilación de histonas y metilación del ADN participan en la etiología de la EA. Las placas amiloides son formadas por el depósito de los péptidos β-amiloides producidos durante el procesamiento de la proteína precursora amiloide (APP) mediante las secretasas β y γ. Interesantemente, durante el procesamiento de la APP, además del fragmento extracelular β amiloide, se produce también un fragmento intracelular llamado dominio intracelular APP (AICD), el cual interactúa in vitro con la HAT-TIP60 y coactúa como un activador transcripcional. Esto sugiere que la EA está asociada con un incremento en la acetilación de histonas18. Tal suposición es apoyada por resultados en cultivos neuronales, los cuales muestran que mutaciones en el gen presenilina1 (PS1) (gen que codifica para un miembro del complejo γ-secretasa) inhiben la degradación vía proteosoma de la proteína HAT-CBP, resultando en un incremento de la expresión génica mediada por CREB20.
En un reciente estudio in vivo se encontró que la sobreexpresión de la histona desacetilasa SIRT1 confiere protección contra la neurodegeneración en un modelo murino para la EA; sin embargo, no se conoce si SIRT1 actúa a través de la maquinaria epigenética y/o a través de otros genes21. Otros estudios han demostrado también una disminución en la acetilación de las histonas, lo cual está causalmente relacionado con la EA22.
Finalmente la metilación del ADN es otro mecanismo implicado en la etiología de la EA, siendo el proceso de hipometilación el más ampliamente reportado. En cultivos celulares la hipometilación de la región promotora del gen PS1 incrementó la expresión de presenilina y potenció la formación de las placas β-amiloides23–26. Se propone que este efecto puede revertirse con la aplicación de un donador de metilo como S-adenosilmetionina (SAM), el cual rescataría la metilación y disminuiría la expresión de presenilina, reduciendo la formación de las placas β-amiloides. Estos estudios sugieren que los donadores de metilo y/o los fármacos que actúan sobre el metabolismo de los metilos pueden ser agentes terapéuticos potenciales para el tratamiento de la EA25–27.
Enfermedad de HuntingtonEs un raro padecimiento neurodegenerativo que se hereda en forma autosómica dominante y cuya incidencia varía de 3-7 por cada 100.000 individuos en los Estados Unidos. Se caracteriza por deterioro cognitivo y de la memoria, alteraciones psiquiátricas y movimientos no coordinados. La EH resulta de una mutación en el gen HTT, consistente en la amplificación del número de repeticiones CAG en el extremo 5′ del gen, lo que produce una expansión de poliglutaminas en la región N-terminal de la proteína huntingtina28.
Varios estudios sugieren que la acetilación y la metilación de las histonas están alteradas en la EH. Modelos murinos en los cuales la actividad de la proteína de unión a CREB (CREBBP) está comprometida exhiben reducida acetilación de la cromatina, disminución de la FLP en el hipocampo y déficit de la memoria a largo plazo. También se ha observado que la pérdida de la función de CREBBP se asocia con degeneración del cuerpo estriado en modelos murinos para la EH. Además, la huntingtina que resulta de la expresión del gen HTT mutado, interactúa directamente con el dominio de las acetiltransferasas CREBBP, resultando en una reducción de la actividad de esta enzima29.
Otro mecanismo observado en cultivos celulares involucra la unión de las poliglutaminas a las proteínas histonas acetiltransferasas (HAT), a CREBBP y al factor asociado p300/CREBBP, lo cual reduce la actividad de las HAT y el nivel de acetilación de las histonas H3 y H4. Estos resultados demuestran que la disminución de los niveles de CREBBP y la alteración en los procesos de transcripción dependientes de CREBBP/CREB1 se asocian con déficit en la memoria a largo plazo en ratones mutantes HdhQ7/Q11129.
En estudios recientes realizados en un modelo de drosophila con expansión de poliglutaminas, se ha logrado reducir la actividad de las HDAC mediante mutaciones dirigidas o inhibición farmacológica, disminuyendo los efectos patológicos inducidos por la poliglutamina29. Actualmente se conoce un grupo amplio de inhibidores de las HDAC, como el butirato de sodio, el ácido suberoyl anilido hidroxalamico (SAHA), el fenilbutirato y el inhibidor 4b de la HDAC, los cuales mejoran el déficit motor y la atrofia neuronal en modelos murinos de EH30–34(fig. 2). Interesantemente, se ha observado reversión del estado de hipoacetilación de la α-tubulina citoplasmática mediante inhibidores de la HDAC20,29. En otro estudio, células estriadas de ratón, con o sin EH, fueron tratadas con el inhibidor específico tubacina HDAC6, obteniendo un incremento en la acetilación de la α-tubulina y reversión de algunos signos de la enfermedad35.

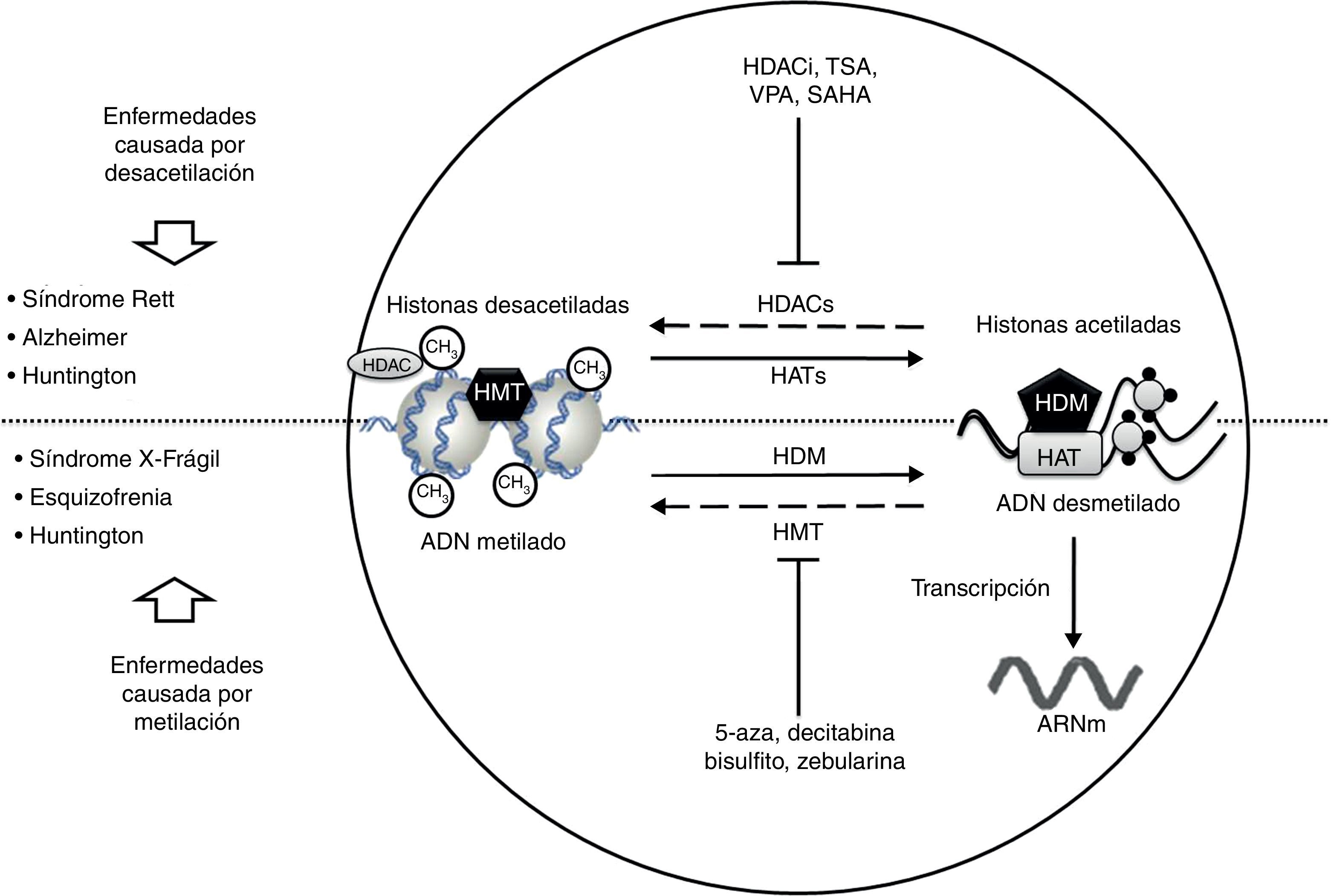
Desórdenes neurológicos y alteraciones epigenéticas. En condiciones normales las enzimas HAT acetilan histonas para una correcta transcripción del ADN; en cambio las enzimas HDAC desacetilan histonas, lo que puede dar lugar a padecimientos como Rett, Alzheimer y Huntington. Sustancias como HDACi, TSA, VPA, SAHA inhiben las enzimas HDAC y son usadas en el tratamiento de dichas patologías.
Por otro lado, en condiciones normales las enzimas HDM desmetilan el ADN, permitiendo su correcta transcripción; en cambio las enzimas HMT metilan el ADN, provocando padecimientos como X-frágil, esquizofrenia y Huntington. Sustancias como 5-aza, decitabina, bisulfito y zebularina inhiben las enzimas HMT y son usadas en el tratamiento de dichas patologías.
HAT: histonas acetiltransferasas; HDAC: histonas desacetilasas; HDACi: inhibidor de histonas desacetilasas; HDM: histonas desmetilasas; HMT: histonas metiltransferasas; SAHA: ácido suberoilsanilidohidroxalamico; TSA: tricostatina A; VPA: ácido valproico; 5-aza: 5- aza-2-deoxicitidina.
Además de la acetilación, también el proceso de metilación se encuentra alterado en modelos murinos de EH, en los que se observa un incremento de la proteína dimetil H3K9, misma que participa en procesos de represión transcripcional25,27,36.
Enfermedades del neurodesarrolloUn número substancial de evidencias señalan qué alteraciones epigenéticas están involucradas en el mecanismo etiológico de varios trastornos del neurodesarrollo; entre ellos los síndromes de Rett (SR), X-frágil (SXF) y la esquizofrenia (tabla 2).
Síndrome de RettTiene una frecuencia de 1:10.000 a 1:15.000 individuos en Estados Unidos. La enfermedad se manifiesta en edades tempranas de la infancia como una alteración del desarrollo neurológico que ocasiona déficit en el aprendizaje y la memoria. Se caracteriza por microcefalia, movimientos estereotipados, alteración de la coordinación motora, convulsiones, problemas del lenguaje y del aprendizaje, y alteración cognitiva de leve a severa. Otras de las manifestaciones clínicas incluyen ataxia, espasticidad y alteraciones autonómicas.
El mecanismo epigenético relacionado al SR es el fenómeno de metilación del ADN que impide la transcripción del gen MECP237. La proteína MECP2 es miembro de la familia de represores transcripcionales de unión a grupos metilo (MBP) y juega un papel dual en el silenciamiento y la activación transcripcional37. Además de regular la metilación del ADN, MECP2 también participa en procesos de acetilación y metilación de histonas (fig. 2).
Los resultados de estudios realizados en modelos murinos con deleciones del gen MECP2 en regiones específicas del cerebro recuerdan el fenotipo de los pacientes con SR38. Un modelo murino carente de la expresión del gen MECP2 mostró cerebros más pequeños, con menor peso y con neuronas más pequeñas; además, los ratones mostraron ausencia total de su actividad exploratoria39,40, déficit cognitivo y reducida plasticidad sináptica41.
Consistente con el efecto de la deficiencia de la proteína MECP2, las alteraciones cognitivas pueden ser revertidas por sobreexpresión del gen MECP227,42,43. La fosforilación de MECP2 también permite el crecimiento dendrítico y la maduración de las espinas dendríticas27,44. En modelos murinos se ha observado que la sobreexpresión de MECP2 induce la FLP en el hipocampo y mejora la formación de la memoria a largo plazo, lo que demuestra que MECP2 modula la inducción de la plasticidad sináptica y la formación de la memoria45.
Síndrome X frágilEs considerado la causa heredable más común de disfunción intelectual ligada al cromosoma X. Su frecuencia es de aproximadamente uno de cada 4.000 varones y una de cada 8.000 mujeres; se asocia a la expresión citogenética del sitio frágil Xq27.3 (FRAXA) y se transmite mediante un mecanismo inusual de herencia ligado al cromosoma X. Además de un conjunto de características dismórficas, los pacientes con SXF presentan deficiencia cognitiva, de aprendizaje y de memoria. La mutación que causa el SXF es una amplificación de trinucleótidos CGG en la región no traducible 5′ del gen FMR119. Actualmente se conoce que la metilación del ADN, así como la acetilación y metilación de las histonas están implicadas en los mecanismos moleculares de esta enfermedad. La expansión del trinucleótido CGG resulta en un incremento de la metilación del ADN en el promotor del gen FMR1, lo cual impide la transcripción génica y anula la producción del ARN mensajero y de la proteína FMR146–48. La proteína FMR1 (FMRP), ausente en los pacientes con SXF, es una proteína de unión a ARNm que participa en la formación de los polirribosomas46.
Recientes estudios en modelos murinos de SXF, demuestran que la regulación de la traducción mediante FMRP es esencial para el aprendizaje y la memoria, tanto en células madre neurales adultas como en neuronas jóvenes; así mismo se ha observado que un reducido número de nuevas neuronas, con defectos en la maduración, puede contribuir a la deficiencia cognitiva del SXF49.
El tratamiento de células linfoblastoides de pacientes con SXF con agentes desmetilantes como la 5-aza-2-deoxycitidina revierte eficientemente la hipermetilación del promotor FMR1, restableciendo los niveles del ARNm50. Interesantemente, la expresión del ARNm es restablecida incluso por encima del nivel de referencia cuando el 5-aza-2-deoxycitidina es combinado con los inhibidores de las HDAC como el 4-fenilbutirato, el butirato de sodio y la TSA51. Esto sugiere un efecto combinado de la desacetilación y la hipermetilación del ADN como mecanismo causal del SXF y, opuestamente, un efecto sinérgico potencial de hiperacetilación de las histonas y desmetilación del ADN como un posible tratamiento para los pacientes con SXF (fig. 2). Dos estudios adicionales reportan que el tratamiento con 5-aza-2-deoxycitidina en líneas celulares de pacientes con SXF desplaza el patrón epigenético de metilación de histonas52. Otros estudios utilizando ensayos de inmunoprecipitación de cromatina en el promotor del gen FMR1 muestran que el tratamiento con 5-aza-2-deoxycitidina disminuye la metilación de la histona H3K9, la cual participa en el silenciamiento transcripcional25,53.
EsquizofreniaEs un padecimiento mental relativamente común, con una prevalencia en los Estados Unidos de 1:100 individuos mayores de 18 años. Se caracteriza por presentar 2 tipos de síntomas psicóticos. Los síntomas positivos incluyen alteraciones en el pensamiento, ilusiones, alucinaciones y delirios. Los síntomas negativos incluyen aislamiento social, pérdida de la motivación, y apatía, reflejando una pérdida de las habilidades conductuales. Presentan además disfunción cognitiva, que incluye el deterioro de la memoria de trabajo y la desorganización conceptual27.
Las causas de la esquizofrenia no son bien entendidas pero es probable que resulten de una compleja interacción de predisposición genética y condiciones ambientales durante el desarrollo pre y posnatal. Las evidencias sugieren el involucramiento de mecanismos epigenéticos, en particular la metilación de histonas y de ADN. En tejido cerebral de pacientes diagnosticados con esquizofrenia, los niveles del ARNm de reelina (RELN) (proteína de la matriz extracelular implicada en la migración neuronal) se encontraron significativamente reducidos, correlacionando con niveles incrementados de la proteína DNA metiltransferasa 1 (DNMT1). Esto sugiere que la hipermetilación del ADN en la región promotora del gen RELN, puede ser responsable de la disminución de los niveles del gen RELN en pacientes esquizofrénicos54. Se ha demostrado que el promotor del gen RELN contiene varios sitios específicos para metilación y que la inhibición de la actividad de HDAC y DNMT incrementa la expresión de RELN, lo que sugiere que mecanismos epigenéticos están importantemente relacionados con la expresión de esta proteína25,45.
Estudios posteriores demuestran que la acetilación de lisinas en la histona 3 (H3K9 o K14), en el promotor del gen RELN en cerebros de murinos se incrementa después del tratamiento con ácido valproico asociado con clozapina o sulpirida55,56. Por otro lado, estudios realizados en células y cultivos neuronales in vivo e in vitro demuestran que la metilación del gen RELN suprime su expresión. El tratamiento con TSA y ácido valproico incrementaron la metilación del ADN en el promotor del gen RELN; en tanto que en ratones heterocigotos para RELN, inyecciones con ácido valproico potencian la expresión del gen RELN. Estos hallazgos sugieren una interacción entre la desmetilación del ADN y la acetilación histónica para la activación de la expresión del gen RELN54.
ConclusionesMecanismos epigenéticos tales como la modificación covalente del ADN y los cambios pos-traduccionales de las histonas se han establecido como reguladores necesarios de la fisiología sináptica y de la memoria. Finalmente se ha determinado que los mecanismos epigenéticos juegan un papel central en las funciones cerebrales, por lo que cualquier alteración puede conducir a trastornos en el desarrollo neurológico o a procesos neurodegenerativos. Los estudios en modelos murinos patológicos han generado la mayor parte del conocimiento que actualmente disponemos al respecto y nos muestran un escenario promisorio con una serie de tratamientos potenciales como los inhibidores de las HDAC en la terapia de algunos padecimientos neurológicos. Sin embargo, y a pesar de la creciente evidencia científica relacionada con el papel que estos mecanismos epigenéticos juegan en los procesos de modificación sináptica, aún existe una enorme cantidad de preguntas no resueltas respecto a estos mecanismos y su control sobre la formación y desarrollo de la memoria a largo plazo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

