



Se comentan dos casos clínicos y se describe la utilidad de los métodos de imágenes en el diagnóstico prenatal de malformaciones así como del apoyo a procedimientos invasivos. Las técnicas de imágenes y en especial las de biología molecular, han tenido un sorprendente avance en los últimos años, con lo que el diagnóstico preciso incluido la del gen alterado, es posible realizarlo antes del nacimiento. En el futuro cercano será posible realizar in utero y de manera no invasiva, el diagnóstico de muchas otras enfermedades que se expresan en el período perinatal.
Two clinical cases and the utility of diagnostic images are described. This techniques and molecular biology have advanced importantly during last decades. In next years, new abnormal genetic conditions as well as many other diseases expressed in the perinatal period, will be diagnosed with non invasive procedures during intrauterine life.
Las malformaciones congénitas tienen una prevalencia aproximada de 2–4% en los recién nacidos (1) y son responsables del 40% de las muertes perinatales (2). Son además una causa importante de morbilidad y representan un elevado costo para su tratamiento y rehabilitación.
Los métodos de imágenes, en especial el ultrasonido, representan la herramienta más importante en el diagnóstico de malformaciones congénitas (3). En la última década, otros métodos de imágenes como resonancia magnética, TAC helicoidal y las técnicas de biología molecular, se han incorporado como métodos diagnósticos de malformaciones durante el período prenatal (4).
Se presentan dos casos clínicos que ilustran el estado actual de las técnicas utilizadas en el diagnóstico prenatal.
Caso 1Paciente de 38 años, multípara de uno, sin antecedentes personales o familiares, cursando embarazo de 23 semanas, consulta para segunda opinión por craneosinostosis fetal.
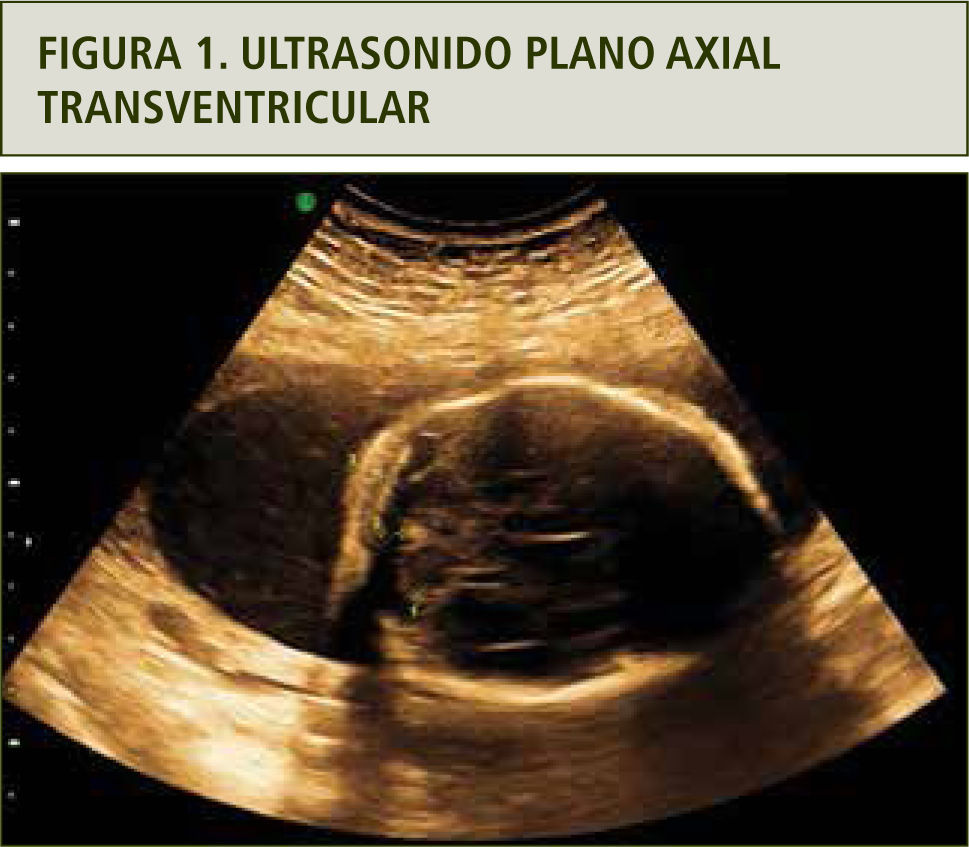
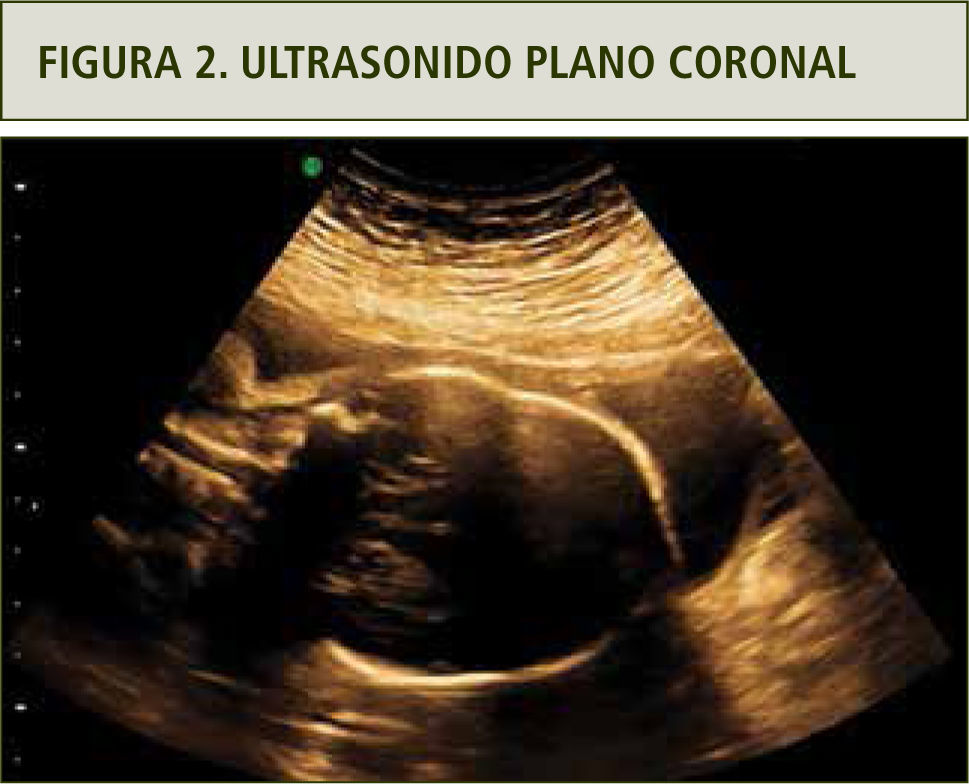
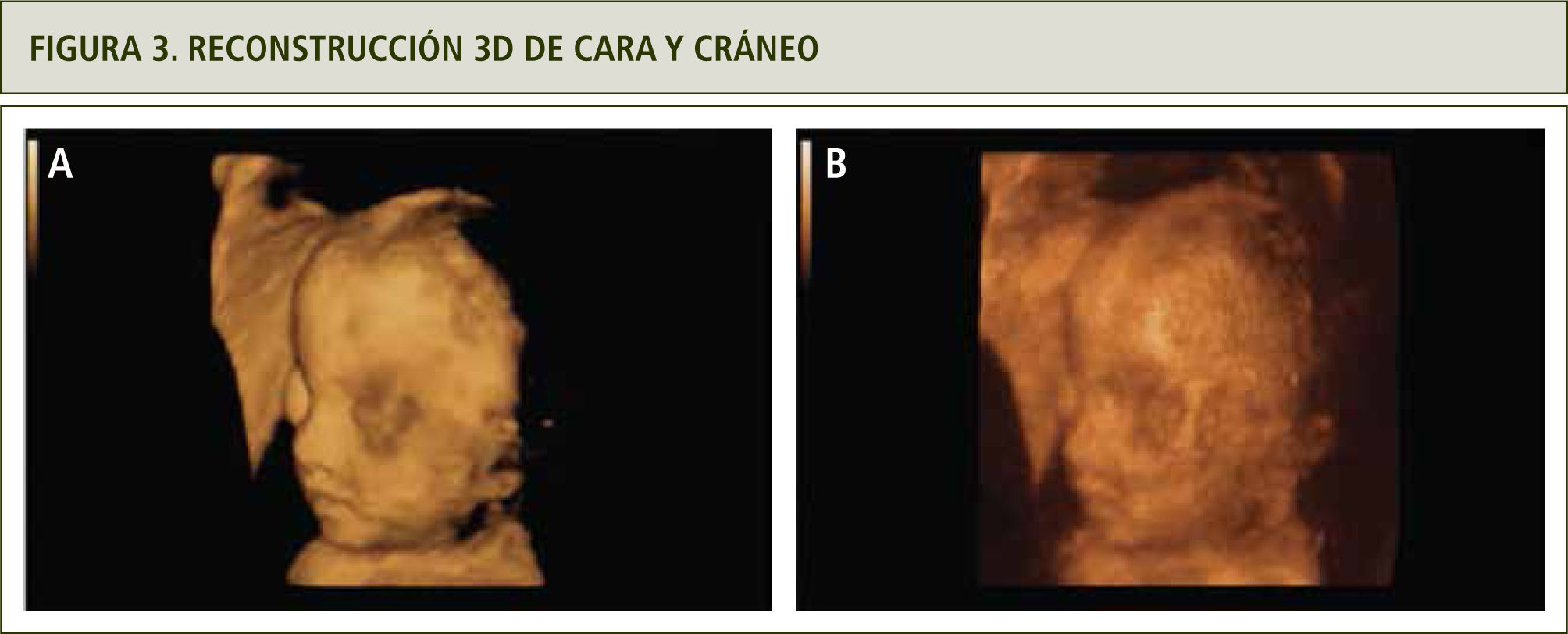
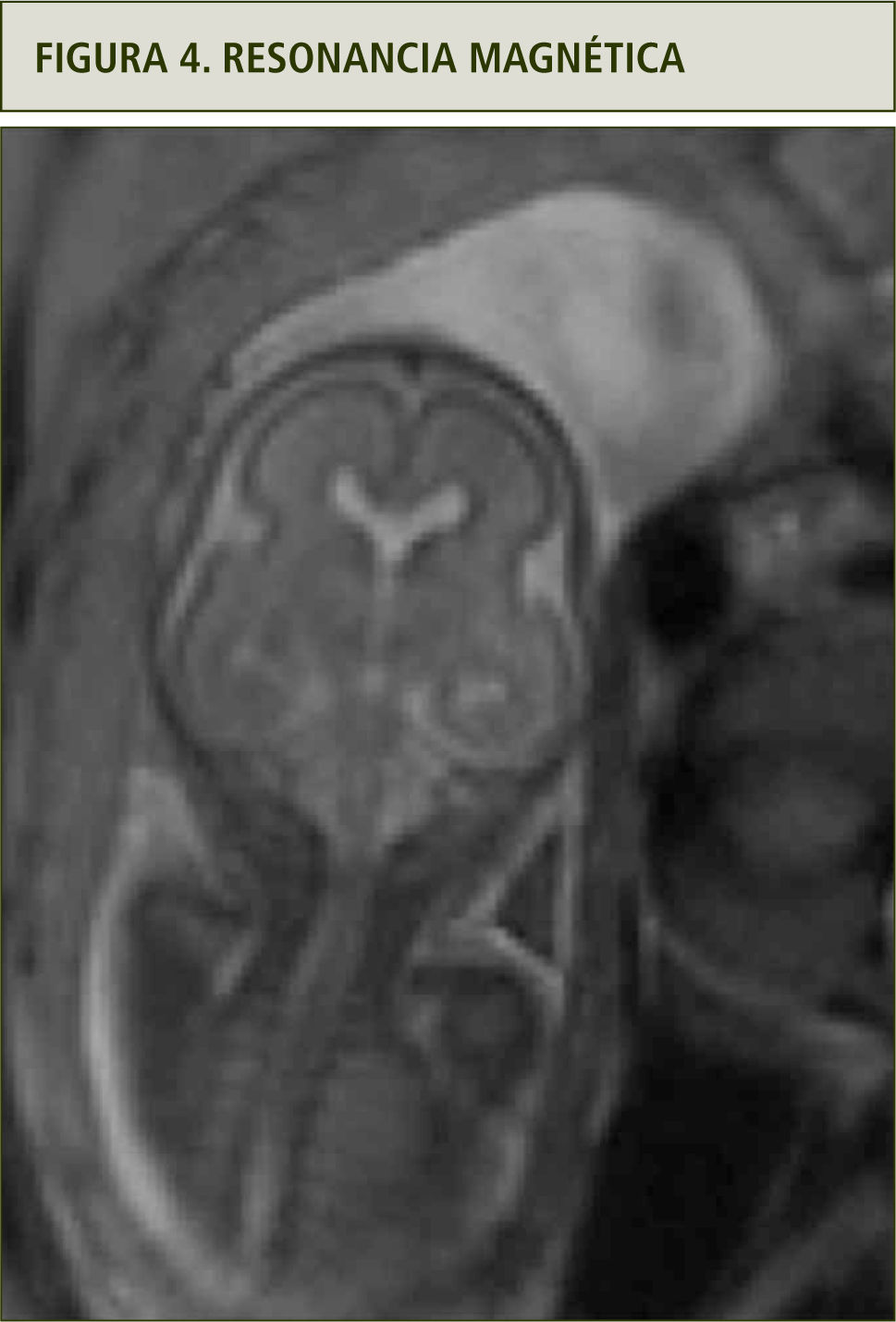
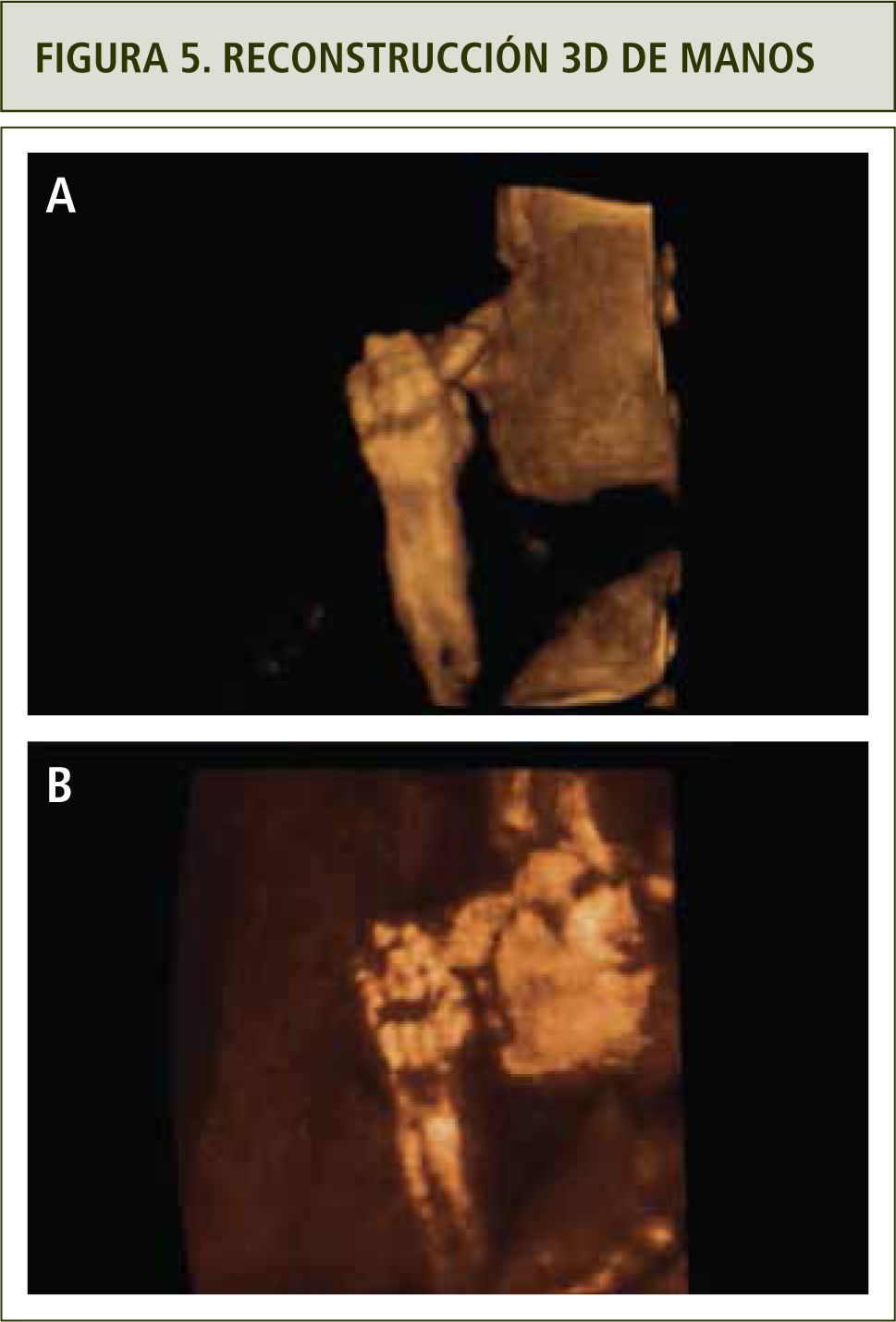
La evaluación ultrasonográfica muestra una leve asimetría del cráneo en planos axiales y coronales, sugerentes de craneosinostosis coronal (figuras 1 y 2). La reconstrucción 3D (figura 3a y b) y la resonancia magnética confirmaron los hallazgos (figura 4). En ambas extremidades se encontraron fusión de los dedos compatibles con sindactilia (figura 5 a y b), todos hallazgos sugerentes de Síndrome de Apert.


 Modo de superficie, se observa cara con depresión de la parte media y ptosis palpebral, ausencia de suturas coronales. b) Modo de rayos x, ausencia de suturas coronales.")

 Modo de superficie, se observa sindactilia. b) Modo de rayos x, sindactilia.")
Se realizó amniocentesis para diagnóstico molecular de craneosinostosis que reveló mutación del gen FGFR2, compatible con Síndrome de Apert.
El Síndrome de Aper, también llamado acrocéfalo sindactilia, se caracteriza por turricefalia debido al cierre prematuro de las suturas coronales, frente prominente y braquisindactilia de severidad variable de manos y pies (5). En el 10% de los casos puede estar asociado a otras malformaciones (6–9) y se origina por una mutación del gen que codifica el receptor del factor de crecimiento fibroblástico (FGFR2) (10).
El diagnóstico diferencial incluye otras enfermedades relacionadas con diferentes mutaciones del gen FGFR2 y que se presentan también con craneosinostosis. Entre éstas se incluyen los síndromes de Pfeiffer, de Crouzon y de Carpenter (11).
El diagnóstico molecular de esta condición se puede realizar mediante el estudio de la mutación del gen FGFR2 en ADN fetal (12).
El pronóstico de esta enfermedad está asociado a un 50% de riesgo de retardo mental en grado variable (13). En el largo plazo se puede presentar obstrucción de la vía aérea alta y dificultades motoras asociadas a la sindactilia. La cirugía plástica juega un importante rol en la reparación de las dismorfias faciales.
En cuanto al riesgo de recurrencia, si bien la mayoría de los casos son esporádicos tiene una herencia autosómica dominante con una probabilidad de hijos afectados de un 50% (14).
Caso 2Paciente de 29 años multípara de uno, cursando gestación de 38 semanas, es referida para resolución del embarazo con diagnóstico de atresia esofágica y polihidroamnios.
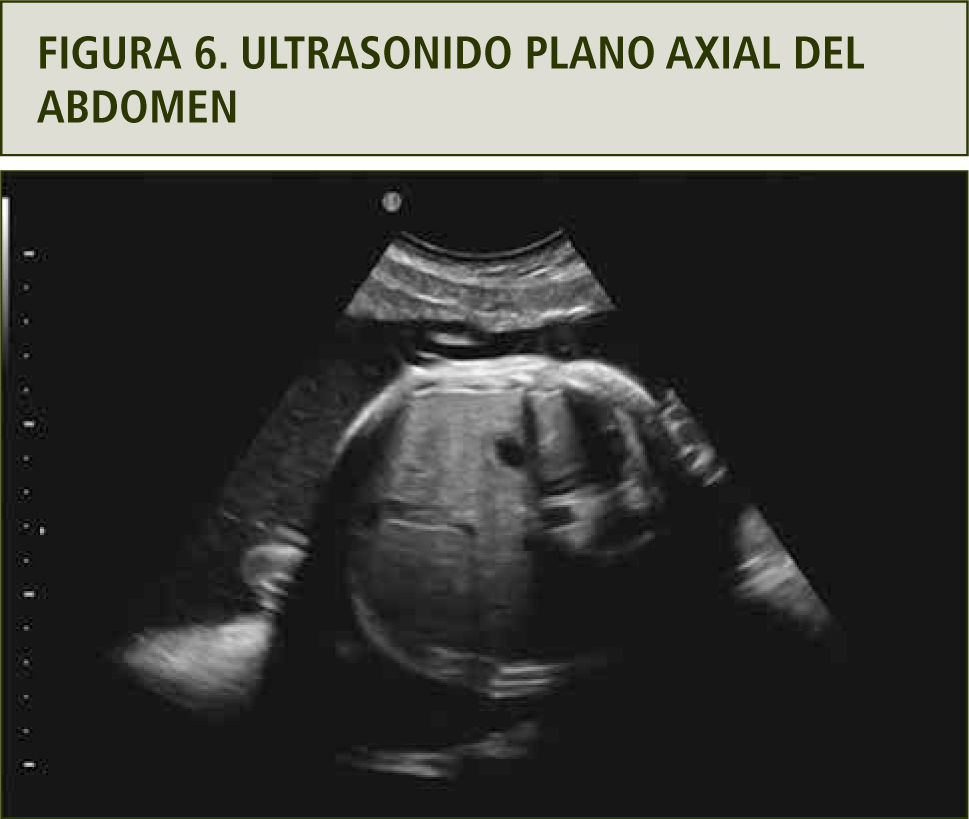
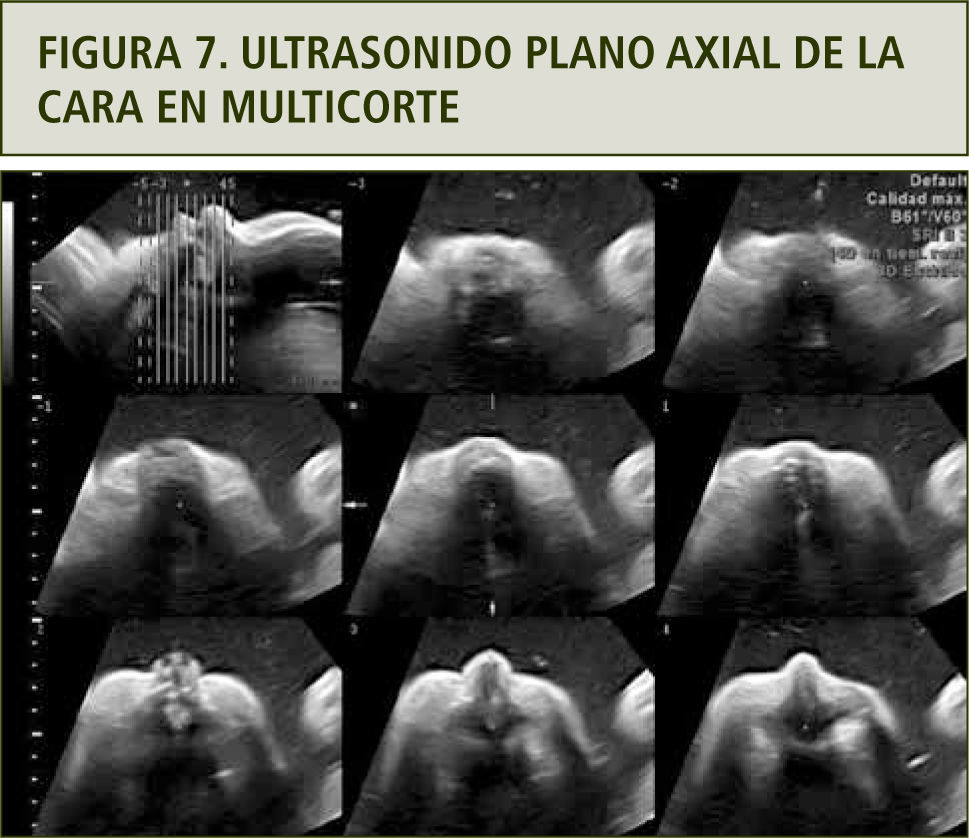
Se realizó evaluación ultrasonográfica que confirmó polihidroamnios y estómago de pequeño tamaño, sin otras alteraciones morfológicas. Esto sugiere una probable atresia esofágica con fístula y vía aérea superior normal (figuras 6 y 7). El cariograma mediante cordocentesis fue normal.


Se realizó interrupción del embarazo con recién nacido sin malformaciones aparentes y con pasaje adecuado de sonda nasogástrica, lo que descartó atresia esofágica.
El RN evolucionó en malas condiciones con insuficiencia respiratoria que requirió ventilación mecánica con buena respuesta evolucionando con bajos requerimientos de oxígeno, por lo que se intentó desconexión sin éxito ya que volvió a caer en insuficiencia respiratoria. Este hecho llevó a plantear una probable obstrucción de la vía aérea superior.
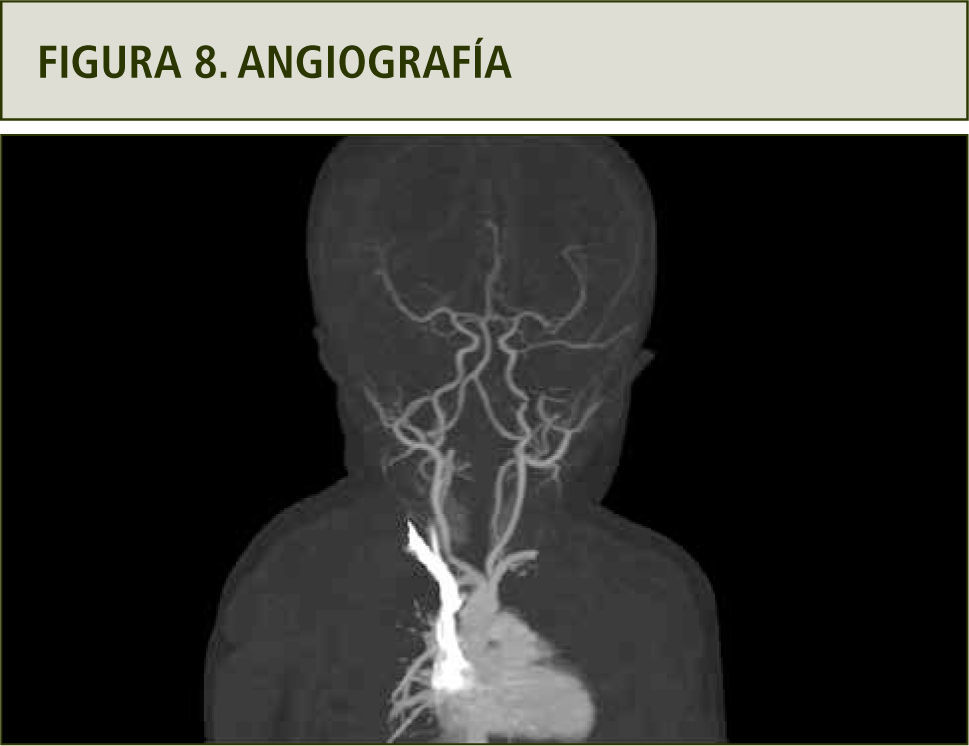
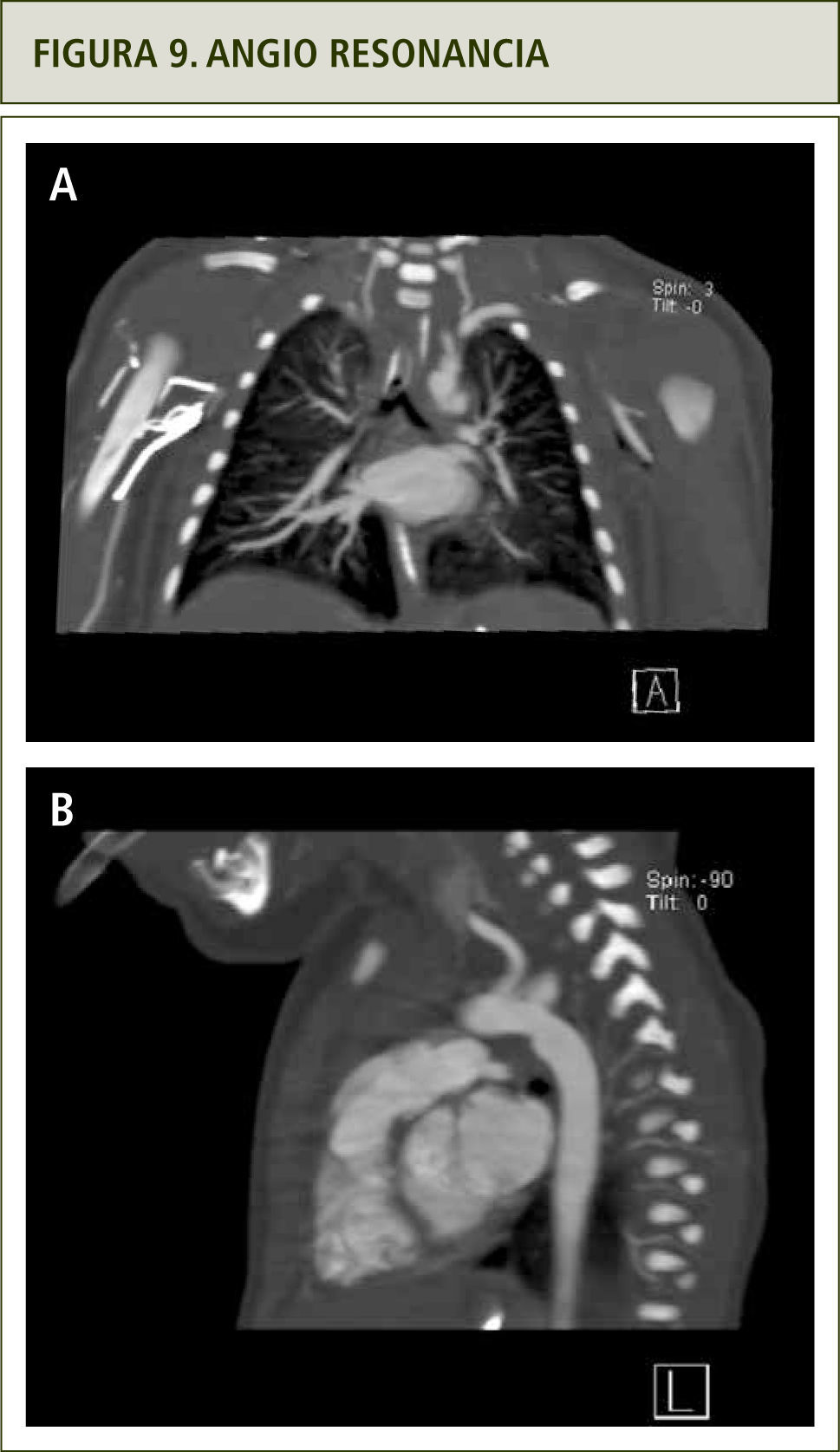
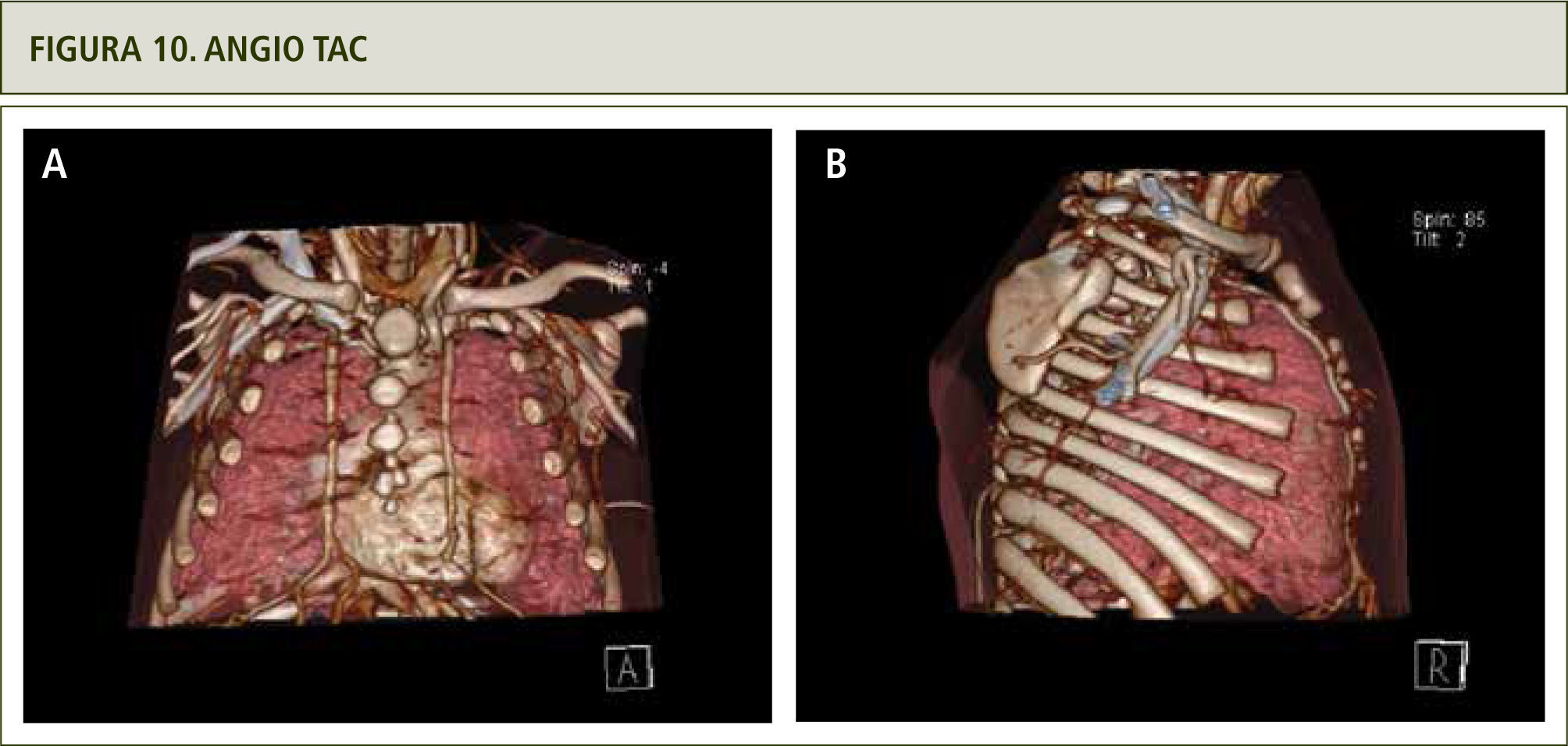
La evaluación con angiografía (figura 8), angio resonancia (figura 9 a y b) y TAC helicoidal descartaron la obstrucción de vía aérea superior, mostrando en cambio que los vasos del cuello y la arteria aorta son tortuosos así como un aumento de tamaño de las arterias mamarias externas (figura 10 a y b). Todos estos hallazgos hicieron plantear el Síndrome de Vasos Tortuosos; una rara enfermedad caracterizada por dilatación, elongación y tortuosidad de los vasos sanguíneos arteriales de mediano y gran tamaño (15). Se asocia además con otras dismorfias que incluyen cara elongada, blefarofimosis, micrognatia y alteraciones del tejido conectivo como piel extensible, híper laxitud de extremidades y aracnodactilia (16).

 Plano coronal de tórax, vasos del cuello y aorta se observan tortuosos. b) Plano sagital de tórax, se observa aorta tortuosa.")
 Visión frontal. b) Visión lateral.")
Estos hechos están causados por una fragmentación de la membrana elástica interna y de las fibras elásticas de la túnica media de las arterias de gran tamaño (17,18).
El origen de esta enfermedad está dado por una mutación del gen SLC2A10, que codifica para el transportador de glucosa GLUT 10 (19) lo que genera un aumento de decorina que, a su vez, inhibe a TGFB, provocando una up regulation de TGFB, que resulta en una inhibición de la matriz extracelular en especial de la elastogénesis (20). El gen SLC2A10 está situado en el cromosoma 20 q13 (21).
El diagnóstico de esta infrecuente condición, se sospecha frente a polihidroamnios y la ausencia de alteraciones metabólicas, malformaciones digestivas o del sistema nervioso central y se objetiva con resonancia magnética o angio TAC que muestra la presencia de vasos tortuosos.
El diagnóstico diferencial incluye otras enfermedades con vasos tortuosos y aneurismas como el Síndrome de Loeys-Dietz y algunas formas recesivas de cutis laxa(22–25).
El pronóstico es reservado ya que los pacientes tienen tendencia a la formación de aneurismas, disección de grandes vasos y alteraciones isquémicas, lo que determina una sobrevida menor del 60% a 5 años (26). Esta enfermedad de condición autosómica recesiva tiene un riesgo de recurrencia de 25% (15,16).
Las técnicas de imágenes y en especial las de biología molecular, han tenido un sorprendente avance en los últimos años, con lo que el diagnóstico preciso incluido la del gen alterado, es posible realizarlo antes del nacimiento. En el futuro cercano será posible realizar in utero, y en forma no invasiva, el diagnóstico de todas las enfermedades que se expresan en el período perinatal.
Los autores declaran no tener conflictos de interés, en relación a este artículo.