



Las enfermedades cardiovasculares constituyen un importante problema de salud pública al ser la principal causa de morbilidad y mortalidad en el mundo. Por ello, existe la creciente necesidad de tratamientos farmacoterapéuticos más eficaces y seguros. Sin embargo, a pesar de que los médicos prescriben fármacos sobre la base de las características farmacológicas del medicamento y la probabilidad de obtener resultados clínicamente reproducibles, muchos de los fármacos son eficaces sólo entre 25-60% de los pacientes. En este sentido es que la Farmacogenómica, a través del estudio de variantes genéticas de proteínas involucradas en la farmacocinética y farmacodinamia de los medicamentos, persigue maximizar su eficacia y seguridad.
Este trabajo pretende dar una visión general acerca de farmacogenómica cardiovascular y la posibilidad de utilizar, en la consulta clínica, herramientas genéticas para apoyar la decisión farmacoterapéutica, con el objeto de mejorar la respuesta al tratamiento de enfermedades cardiovasculares, un paso hacia la medicina personalizada en Chile.
Cardiovascular disease is a major public health problem being the leading cause of morbidity and mortality worldwide. Therefore, there is a growing need for safer and more effective pharmacotherapeutic treatments. However, although physicians prescribe drugs based on pharmacological properties of each drug and the probability of obtaining clinically reproducible results, many drugs are effective only in 25-60% of patients. In this respect, pharmacogenomics, through the study of genetic variants of proteins involved in the pharmacokinetics and pharmacodynamics of drugs, pursues to maximize their efficacy and safety.
This paper aims to give an overview of cardiovascular pharmacogenomics and the possibility to use, in clinical practice, genetic tools to support pharmacotherapeutical decisions, in order to improve the response to treatment of cardiovascular diseases, a step toward personalized medicine in Chile.
Las enfermedades cardiovasculares constituyen un problema de salud pública porque son la principal causa de morbilidad y mortalidad en todo el mundo (17,5 millones de muertes/año)1. En Chile, las enfermedades cardiovasculares también representan la primera causa de muerte desde el año 1969. En el año 2011 la tasa de mortalidad en Chile era de 149,3 por 100.000 habitantes (25.744 defunciones anuales)2. Asimismo, al analizar por grupos específicos de causas de mortalidad destacan la mortalidad por enfermedad isquémica del corazón, con una tasa de 41,7 por 100.000 habitantes y las enfermedades cerebrovasculares, con una tasa de 50,6 por 100.000 habitantes2.
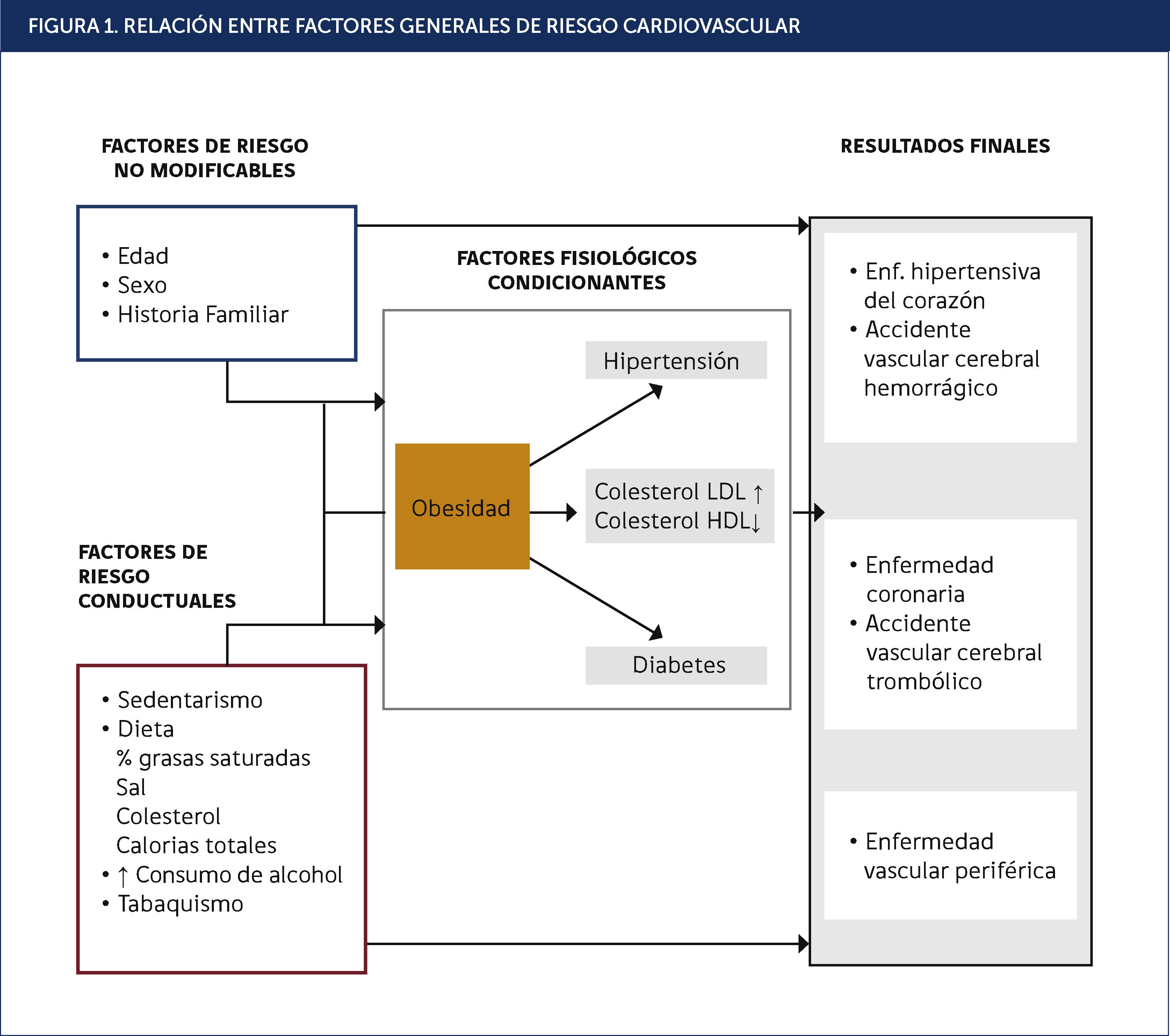
Diversos factores ayudan a que se produzcan estas enfermedades cardiovasculares. Existen factores de riesgo no modificables como la herencia genética y otros modificables tales como el hábito de fumar, la ingesta de alcohol, la alimentación y la actividad física. Estos últimos son determinantes en el desarrollo de la obesidad, que es uno de los principales factores desencadenantes de otros factores de riesgos mayores, denominados fisiológicos, tales como la diabetes, la hipertensión arterial y el niveles de colesterol sanguíneo. Cuando una persona tiene más de uno de estos factores de riesgo, aumentan sus probabilidades de padecer una enfermedad. El nivel de riesgo global de un individuo es el que determina la probabilidad de hacer una complicación cardiovascular, tales como el infarto agudo al miocardio, ataque cerebral, entre otros (Figura 1)3, aunque hay que reconocer que la mayoría de los eventos cardiovasculares en una población ocurre en personas con niveles promedio o ligeramente elevados de los factores de riesgo4.

La evidencia que han entregado tres estudios clínicos, “Major Cardiovascular Risk Factors in Latin America: A Comparison with the United States. LASO 2013”5, “The Global Burden of Disease Study (GBD) 2010”6 y “Cardiovascular risk awareness, treatment, and control in urban Latin America”7, muestra que dentro de uno de los principales factores de riesgo cardiovascular prevalentes, se encuentra la hipertensión, y en conjunto con un alto nivel de colesterol y bajo HDL. Al respecto, en Chile se han introducido nuevas políticas públicas, con el objeto de disminuir los eventos cardiovasculares. Estas están enfocadas principalmente acciones preventivas, las cuales se dirigen a intervenir la población general (estrategia poblacional)8•12 y pacientes (estrategia de alto riesgo)13, disminuyendo la hipertensión, el nivel de colesterol-LDL y aumentando el nivel de HDL.
A partir del año 2005 con las Garantías Explícitas en Salud (GES), asegurando el acceso, oportunidad y protección financiera a los beneficiarios de varias patologías, incluyendo aquellas condiciones que aumentan el riesgo cardiovascular10, además de metas de cobertura para exámenes de medicina preventiva para intervenciones oportunas, por ejemplo la detección de los factores de riesgo cardiovascular mayores, se ha contribuido a asegurar el diagnóstico, tratamiento y seguimiento de pacientes beneficiarios del sistema público y privado.
La prevención y tratamiento eficaces de las enfermedades cardiovasculares requieren exámenes regulares para los factores de riesgo, un alto conocimiento de la enfermedad, un tratamiento eficaz de los factores de riesgo identificados y la adherencia al tratamiento prescrito.
Una serie de medicamentos, entre los que se incluye: antiarrítmicos, anticoagulantes, beta-bloqueadores, bloqueadores de calcio, inhibidores de receptores de angiotensina, digitálicos, diuréticos, inhibidores de la enzima convertidora de la angiotensina (ECA), constituyen el arsenal farmacoterapéutico disponible. El medicamento a elegir por el médico será de acuerdo a las características de cada paciente y considerando las recomendación de las Guías Clínicas14.
FARMACOGENÓMICAEl éxito de la medicina moderna es en parte el resultado de un tratamiento farmacológico eficaz. Un hecho bien conocido es que los individuos responden en forma diferente frente a una terapia de fármacos y que ningún medicamento es 100% eficaz en todos los pacientes. En consecuencia, el margen de respuesta a un tratamiento farmacológico es diverso, ya que algunos individuos pueden obtener los efectos esperados, mientras otros no obtienen un resultado terapéutico e incluso pueden experimentar efectos adversos15•17.
La existencia de una heterogeneidad interindividual en respuesta a los fármacos, que afecta tanto a la eficacia como la toxicidad, puede ser mediada a través de la alteración de la farmacocinética y farmacodinámica de los medicamentos. Estos mecanismos de variabilidad están moldeados por la interacción genético-ambiental. La contribución de cada factor varía con cada fármaco18,19.
La relación entre las reacciones adversas de fármacos y las variaciones determinadas genéticamente fue demostrada por primera vez en los años 50. Friedrich Vogel fue el primero en usar el término farmacogenética en 1959, pero no fue hasta 1962 cuando la farmacogenética fue definida como el estudio de las variaciones genéticas que causan la variabilidad en la respuesta a los fármacos20. Los estudios de farmacogenética se basan en la investigación de genes candidatos seleccionados por su importancia biológica, ya sea en la cinética o por su relación en la acción farmacológica; el objetivo final es identificar individuos con riesgo de experimentar efectos adversos o con probabilidad de ser resistentes al tratamiento.
De acuerdo a lo anterior, este trabajo busca dar una visión general acerca de la posibilidad de utilizar y aplicar en la consulta clínica diaria herramientas genéticas para mejorar la decisión terapéutica, con el objeto de mejorar la eficacia del tratamiento enfermedades cardiovasculares.
APLICACIONES DE LA FARMACOGENÓMICA Y FARMACOGENÉTICA EN LA CLÍNICATras el éxito del Proyecto Genoma Humano, el Proyecto Internacional HapMap y el Proyecto 1.000 Genomas, nuestra comprensión de la variación genética humana ha aumentado de forma secuencial. Investigaciones extensas concomitantes se han llevado a cabo para encontrar biomarcadores genéticos asociados con la susceptibilidad, el diagnóstico, el pronóstico de la enfermedad y la respuesta al tratamiento con técnicas de genes candidatos, estudios de asociación del genoma completo (“Genome-Wide Association Studies”, GWAS) y de nuevas generación de tecnologías de secuenciación.
Es interesante observar que estudios de genes candidatos farmacogenómicos han sido sustancialmente más exitosos en la identificación de las variantes comunes replicables de tamaño del efecto apreciable en comparación con las investigaciones de genes candidatos en la genética de la enfermedad. Esto es posiblemente debido a una mayor comprensión de las vías farmacológicas en comparación con los procesos de enfermedad21.
Además, aunque a gran escala GWAS y posterior meta-análisis22•24 están ahora descubriendo asociaciones genéticas de la susceptibilidad a las enfermedades cardiovasculares comunes, los tamaños del efecto variante son, en su mayoría inferiores a los de las asociaciones de farmacogenéticas, particularmente los relacionadas con reacciones adversas (RAMs)25•28.
Sin embargo, a pesar de las altas expectativas, la transferencia de las asociaciones genéticas a la práctica clínica ha sido lenta, habiendo algunas excepciones notables. Es el caso de la identificación del genotipo HLA-B*57:01 antes de administrar la terapia antirretroviral abacavir, debido a que se ha demostrado que la presencia de esta variante aumenta significativamente la incidencia de síndrome de hipersensibilidad al medicamento29. Sin embargo, este es un fenotipo de seguridad, mientras que en la enfermedad cardiovascular, la mayoría de asociaciones farmacogenómicas se han centrado en los puntos finales de eficacia, donde la magnitud del efecto es mucho menor y por lo tanto, aún ninguna asociación validada drogas/gen.
Otra excepción notable se encuentra en el ámbito de la oncología, donde hay un creciente arsenal de terapias genotipo-dependiente con licencia. Por ejemplo, el tamoxifeno está indicado para la prevención de la recurrencia de la enfermedad sólo en los pacientes con cáncer de mama receptores de estrógenos positivos y es anterior a cualquiera de los avances genómicos que se han producido en este siglo. Más recientemente vemurafenib que inhibe BRAF E600 (mutación positiva), pero no el tipo silvestre (BRAF V600) ha sido aprobado para el tratamiento del melanoma no resecable o metastásico.
Los estudios de farmacogenética en cardiología se han centrado en gran medida en los medicamentos que ya tienen licencia y de uso clínico generalizado. Esto, sin duda, representa un obstáculo adicional, porque cambiar la práctica clínica aceptada, y por lo tanto, el comportamiento del médico, es difícil, ya que a menudo requiere un mayor nivel de evidencia.
A continuación se describen algunos esfuerzos farmacogenómicos en medicamentos de uso cardiovascular:
Anticoagulantes: warfarina, acenocumarol y clopidogrelWarfarina y el acenocumarol son los anticoagulantes orales derivados cumarínicos más prescritos en todo el mundo30, y están indicados en pacientes con tromboembolismo venoso, fibrilación auricular y válvulas cardíacas mecánicas. Los fármacos se administran en forma de mezclas racémicas que constan de 50% de cada enantiómero. Son antagonistas de la vitamina K epóxido reductasa (Figura 2). Aunque el mecanismo de acción de estos fármacos es similar, hay algunas diferencias importantes en su farmacocinética, por ejemplo warfarina presenta menor actividad que la acenocumarol y mayores RAMs31,32.
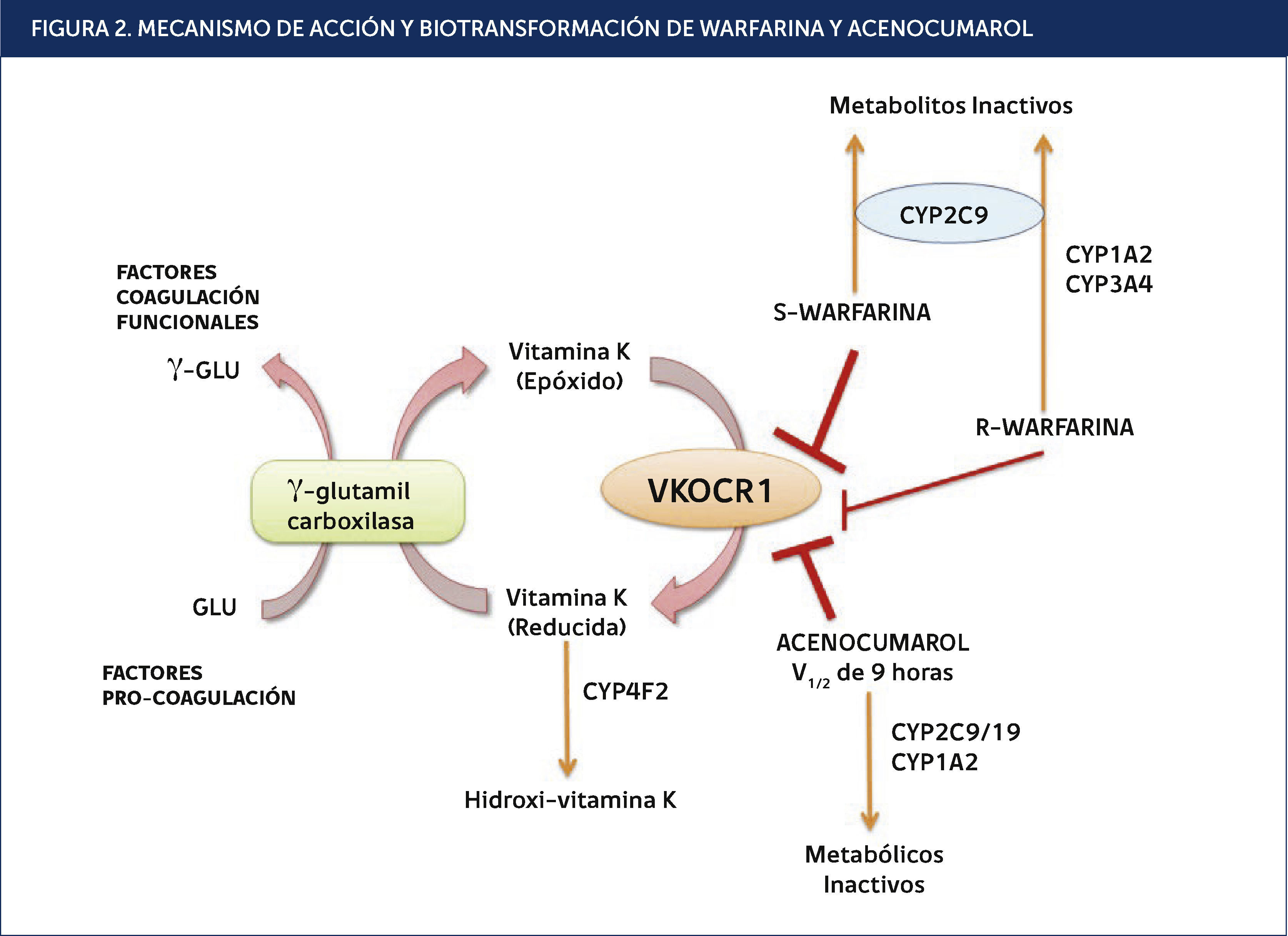
 y el Acenocumarol inhiben la reductasa de VKORC1. Estos antagonizan la regeneración de la reducción de la vitamina K, que es el cofactor esencial para la glutamil carboxilasa. Esto disminuye la activación de la traducción carboxilación de residuos de glutamato en los factores de coagulación II, VII, IX y X. El más potente estereoisómero S-warfarina se inactiva por el CYP2C9, mientras R-warfarin se metaboliza a alcoholes inactivos por CYP1A1, CYP1A2 y CYP3A4, Acenocumarol se inactiva por CYP1A2 y CYP2C9/19. CYP4F2 agota la vitamina K reducida. VKORC1: Vitamina K Epóxido Reductasa Subunidad Complejo 1. Adaptada de Miranda et al., 2011.")
. MECANISMO DE ACCIÓN Y BIOTRANSFORMACIÓN DE WARFARINA Y ACENOCUMAROL
La warfarina (mezcla racémica de dos enantiómeros) y el Acenocumarol inhiben la reductasa de VKORC1. Estos antagonizan la regeneración de la reducción de la vitamina K, que es el cofactor esencial para la glutamil carboxilasa. Esto disminuye la activación de la traducción carboxilación de residuos de glutamato en los factores de coagulación II, VII, IX y X. El más potente estereoisómero S-warfarina se inactiva por el CYP2C9, mientras R-warfarin se metaboliza a alcoholes inactivos por CYP1A1, CYP1A2 y CYP3A4, Acenocumarol se inactiva por CYP1A2 y CYP2C9/19. CYP4F2 agota la vitamina K reducida. VKORC1: Vitamina K Epóxido Reductasa Subunidad Complejo 1. Adaptada de Miranda et al., 2011.
La enzima Vitamina K Epóxido Reductasa Subunidad Complejo 1 (VKORC1), blanco terapéutico de estos medicamentos, está encargada de la regeneración de la reducción de la vitamina K, que es el cofactor esencial para la glutamil carboxilasa. Esta enzima es polimórfica, presenta un polimorfismo de un solo nucleótido no codificante (SNP), denominado rs9923231 (-1639G A; G3673A), en el que se altera un sitio de unión en la región promotora de VKORC1 y produce una disminución expresión30. Como consecuencia de ello se requieren menores dosis de warfarina33. Esto se ha confirmado en varias poblaciones incluyendo caucásicas y africanos, asiáticos34. Sin embargo, VKORC1 rs9923231 sólo explica 20-25% de la variación en la dosis de mantenimiento de warfarina en poblaciones caucásicas y asiáticas, y el ∱/46% de la variabilidad de la dosis en los afroamericanos, lo que es atribuible a la menor frecuencia de sus alelos21,30,35.
Varios autores han demostrado que la dosis de acenocumarol también está influenciada por el genotipo VKORC1. Reitsma y sus colaboradores en el año 2005 ya habían demostrado que los pacientes holandeses que llevan uno o dos alelos variantes para el polimorfismo 1173 requieren una dosis 28% y 47% menores, respectivamente, en comparación con los tipos silvestres36. En Griegos, los portadores heterocigotos de un alelo variante requieren una dosis 19% más baja y en homocigotos una dosis 63% inferior37. Porcentajes similares fueron encontrados en poblaciones de Alemania, Austria, Serbia y el Líbano38•40.
La enzima P450 CYP2C9 metaboliza el enantiómero más potente de S-warfarina y más de 30 variantes alélicas son reconocidas en esta enzima. CYP2C9*2 (rs1799853) y CYP2C9*3 (rs1057910) son las variantes genéticas de CYP2C9 más frecuentes en caucásicos, con frecuencias de 0,13 y 0,07 respectivamente30. En los asiáticos, CYP2C9*2 es muy raro y la frecuencia de CYP2C9*3 es 0,0430. CYP2C9*2 y CYP2C9*3 codifican proteínas cuyas actividades enzimáticas está reducida en un 30-40% y 80-90%, respectivamente30. De acuerdo a ello, están asociadas con una prolongada vida media warfarina, aumentando el tiempo para alcanzar niveles terapéuticos de INR, requiriendo de una reducción de la dosis de warfarina30, y para los pacientes con CYP2C9*3, un mayor riesgo de hemorragia ha sido confirmada25. Los cocientes de riesgo reportados por sangrado en pacientes con genotipo CYP2C9 *1/*3 y *3/*3 en comparación con el genotipo silvestre *1/*1 es de 2,05 (95%, IC=1,36-3,10) y 4,87 (95%, CI=1,38-17,14), respectivamente25. En general, el genotipo CYP2C9da cuenta del 7-10% de la variabilidad de la dosis de warfarina21.
Las enzimas involucradas en el metabolismo de acenocumarol son CYP2C9, CYP1A2 y CYP2C1931. Aunque poco se ha publicado sobre los genotipos CYP2C9 y dosis de acenocumarol comparado con warfarina, hay varios estudios que confirman las asociaciones encontradas con la warfarina, genotipos y el riesgo de sangrado durante acenocumarol. La presencia de un alelo CYP2C9*3 reduce el metabolismo del normalmente inactivo S-acenocumarol y por lo tanto aumenta la vida media de éste enantiómero41. Significa por lo tanto, que el paciente requiere de dosis 19-29% menor de acenocumarol en los portadores de este alelo que en genotipos silvestres42, pero también 13-15% menor en portadores de un alelo *243,44. El riesgo de sobre-anticoagulación y hemorragia grave se incrementa en portadores de la variante *342•45.
Investigaciones previas para acenocumarol demuestran que CYP2C9 representa el 14% de la variabilidad interindividual en la respuesta farmacológica31,32, por lo que no es posible establecer una correlación entre las concentraciones plasmáticas de acenocumarol y el nivel de protrombina o INR. Sin perjuicio de lo anterior, resultados preliminares de nuestro grupo muestran que la variante genética CYP2C9*2, cuando está presente de manera homocigota (*2/*2) se asocia a variaciones en los niveles plasmáticos de acenocumarol y otras variables farmacocinéticas46. Por lo tanto, la importancia de las variantes genéticas de CYP2C9 en la variabilidad en la respuesta a acenocumarol no debe ser subestimada.
Los primeros algoritmos de dosificación para warfarina que incorporan genotipo CYP2C9 fueron publicados en 200447•49. El algoritmo por Gage et al. fue el más extenso y se incluye, además de genotipo CYP2C9, edad, superficie corporal, el sexo, la raza, el INR, el uso de amiodarona y simvastatina. El algoritmo explica el 39% de la variación en la dosis diaria de warfarina. Desde entonces, más de 30 algoritmos se han publicado sobre la base de genotipos de CYP2C9 y VKORC1. Sconce et al. ha publicado uno de los primeros algoritmos, incluyendo CYP2C9 y VKORC1 genotipos así como la edad y la altura50.
En 2008, Gage et al. publicó un algoritmo actualizado incluyendo CYP2C9 y VKORC1 genotipo, edad, superficie corporal, el uso de amiodarona, INR, la raza y la condición de fumador51. En una población caucásica este algoritmo explicó 57% de la variación de la dosis, pero el valor predictivo fue menor (31%) en los afroamericanos. Este tipo de algoritmos ha sido adoptado por la FDA y actualmente esta entidad recomienda su uso.
Wadelius et al. fueron capaces de explicar casi el 59% de la variación en una población sueca, utilizando la información en ambos genotipos, edad, raza, sexo y el número de interactuar fármacos capaces de aumentar el INR52. El R2 univariado de genotipo CYP2C9 fue de aproximadamente 12% y la de VKORC1 29%. La warfarina Farmacogenética Consorcio Internacional (IWPC) creó un algoritmo en una población más diversa de nueve países en cuatro continentes53. 47% de la variación de dosis se explica por CYP2C9, VKORC1, edad, altura, peso, uso de amiodarona, la raza y el número de inductores enzimáticos CYP.
Para warfarina, muchos más algoritmos han sido publicados en diferentes poblaciones de varios países, la mayoría de estos estudios han incluido genotipos VKORC1 y CYP2C9, pero algunos también han incluido CYP4F2, CCCG y genotipos APOE. Las fórmulas de estos estudios han permitido calcular un mantenimiento la dosis de warfarina. Sin embargo, sólo un puñado de estudios han analizado los algoritmos para otros tipos de dosis de cumarinas. Cuando un paciente inicia la warfarina en una dosis farmacogenética-guiada, es difícil saber cómo ajustar esta dosis después de la medición del INR54.
Van Schie y sus colaboradores desarrollaron un algoritmo genotipo-guiado para acenocumarol en una población holandesa55. Los autores también proporcionan dosis de carga relacionados con la dosis de mantenimiento calculado y validaron el algoritmo de acenocumarol más tarde, el cual explicó un 52,7% de la variabilidad6.
Por su parte, el profármaco oral clopidogrel es una tienopiridina de segunda generación cuyo metabolito activo se une irreversiblemente a los receptores de membrana de plaquetas purinérgicos P2Y12 durante toda la vida de una plaqueta (∱/410 días)56 y antagoniza la agregación plaquetaria mediada por ADP. Su metabolismo es complejo: 85% es rápidamente hidrolizado a un metabolito inactivo por la carboxiesterasa hepática 1 (CES1)57, y el resto se somete a dos etapas de oxidación hepática secuenciales para formar primero el metabolito inactivo intermedio (2-oxo-clopidogrel) y luego el metabolito activo (R-130964) mediante CYP1A2, CYP3A4/5, CYP2B6, CYP2C9 y CYP2C1957.
Hay una variabilidad sustancial en la respuesta plaquetaria a clopidogrel58. Una serie de factores contribuyen a esta variabilidad, entre ellos, la edad avanzada (65 años), el índice de masa corporal (IMC), medicamentos que inhiben enzimas CYP (estatinas, inhibidores de la bomba de protones, eritromicina, etc), patologías como la diabetes mellitus, la insuficiencia renal y la disminución de la función ventricular izquierda, pero juntos, todos estos factores sólo explican una pequeña proporción de variabilidad observada58.
CYP2C19 es mayormente responsable de la conversión de clopidogrel inactivo a su metabolito activo57. CYP2C19*1 es el alelo de tipo silvestre, pero más de 25 variantes han sido identificadas; la mayoría poseen una actividad enzimática reducida y son poco frecuentes, a excepción de CYP2C19*2 (rs4244285, c.681G>A), que junto a CYP2C19*3 se asocian con una reducción en circulación activa nivel del metabolito de clopidogrel59.
Según nuestro conocimiento, 13 meta-análisis se han publicado hasta la fecha desde 2010, los que han evaluado la asociación entre los genotipos de CYP2C19 y los resultados clínicos. Un hallazgo consistente y robusto es que la reducción de la función de los alelos CYP2C19 (predominantemente CYP2C19*2) aumenta significativamente el riesgo de trombosis después de una intervención coronaria percutánea (ICP) en comparación con los no portadores. Además, una tendencia de la dosis génica es evidente: la asociación de llevar uno y dos alelos CYP2C19 con pérdida de función (predominantemente CYP2C19*2) con riesgo de trombosis es evidente y significativa60.
Por otro lado, la enzima CES1, principal enzima responsable de la biotransformación de clopidogrel, 2-oxoclopidogrel y su metabolito activo R-130964 a compuestos inactivos ácidos carboxílicos, posee 39 variantes genéticas que pueden contribuir a la variabilidad interindividual en la respuesta al clopidogrel. Recientes investigaciones in vitro han demostrado que la variante G143E (rs71647871) de la isoforma CES1A1 tiene actividad catalítica completamente disminuida para metabolizar clopidogrel y 2-oxo-clopidogrel57, lo que se ha asociado con mayores niveles de metabolitos activo de clopidogrel y reducción de la agregación de plaquetas estimulada por ADP después en pacientes con enfermedad coronaria. De acuerdo a lo anterior, el efecto de G143E debe ser abordado como un biomarcador genético en el tratamiento anticoagulante relacionado con patologías cardiovasculares.
Otro factor interesante a estudiar es el gen ABCB1 (ATP-binding cassette (ABC) subfamily B (MDR/TAP) member 1), que codifica para una glicoproteína P, el cual es un transportador de eflujo que tiene una amplia especificidad, presentando un papel importante en la eliminación de sustratos en el intestino, la orina y la bilis. Una variante ABCB1 comúnmente estudiada es el 3435C T (rs1045642, Ile1145Ile). Se ha observado que pacientes homocigotos mutados (TT) han reducido la absorción de clopidogrel después de una dosis oral única en comparación con pacientes C/T y CC, aún cuando los resultados de otros estudios muestran controversias al respecto61,62.
Las estatinasLas estatinas, la clase farmacológica más comúnmente recetada en todo el mundo, están indicadas para la prevención primaria y secundaria de la enfermedad cardiovascular. Su principal mecanismo de acción es la reducción de LDL y colesterol a través de inhibición competitiva de la enzima 3-hidroxi-3-metilglutaril-coenzima A (HMG-CoA) reductasa, enzima limitante para la síntesis de novo de colesterol. La variación genética en la eficacia hipolipemiante de las estatinas ha sido ampliamente investigada y más de 40 genes candidatos han sido descritos63. Resultado de estos estudios se ha logrado identificar como factor farmacogenómico relevante al gen SLCO1B1 (Solute Carrier Organic Anion Transporter Family Member 1B1), particularmente las variantes genéticas rs4363657 y rs4149056 (521T C, V174A; SLCO1B1*5), en relación principalmente a la toxicidad muscular inducida por estatinas. La frecuencia de miopatía y rabdomiolisis inducida por estatinas se estima en 1/1000 y 1/100.000, respectivamente26,64,65. Los estudios realizados han informado que existe una fuerte asociación entre la variante y la presencia de miopatía grave. La asociación entre la variante genética y reacciones adversas a estatinas se ha demostrado particularmente con simvastatina. Existe falta de documentación de apoyo acerca de su participación en el tratamiento con pravastatina, rosuvastatina y atorvastatina66•68.
En 201264 y posteriormente ratificado el 201465, el CPIC (Clinical Pharmacogenetics Implementation Consortium) emitió directrices de consenso para SLCO1B1*5 y su relación con miopatía inducida por simvastatina, incluyendo la consideración de reducciones de la dosis o el uso de una estatina alternativa para los pacientes que presenten uno o dos alelos con la variante. Sin embargo, esto no es ampliamente practicado actualmente.
Algunas otras variantes genéticas relativas a la influencia de otros genes no metabolizadores de fármacos de uso cardiovascular pero igualmente relevantes han sido estudiadas por nuestro grupo y establecen la importancia de la variabilidad genética en patologías cardiovasculares de los factores de coagulación II y VL, la enzima convertidora de angiotensina (ACE), la proteína transportadora de ésteres de colesterol (CETP), la apolipoproteína E (ApoE), el inhibidor del activador de plasminógeno (PAI-1) y la metiléntetrahidrofolato reductasa (MTHFR)69.
De acuerdo a ello, el planteamiento de utilizar biomarcadores moleculares con paneles genómicos-pharmacogenómicos se plantea abiertamente como el área de desarrollo futuro para la evaluación de la susceptibilidad a eventos cardiovasculares y respuesta a la farmacoterapia.
DISCUSIÓN Y CONCLUSIONESA pesar de un creciente número de estudios y asociaciones establecidas entre respuesta farmacológica y genes del metabolismo de drogas (farmacogenómica), la mayoría de la investigación cardiovascular de este tipo se encuentra en etapas iniciales, ya que los investigadores han encontrado dificultades para identificar y validar asociaciones, lo que se debe mayormente a la heterogeneidad de las poblaciones de pacientes y sus fenotipos o a la dificultad de disponer de tamaños de muestra adecuados. La creación de consorcios de estudio ha permitido abordar estas limitantes y avanzar hacia la normalización de las investigaciones, el mejor uso de recursos financieros involucrados, la realización de estudios más amplios, la evaluación de las asociaciones de farmacogenómicas en diferentes grupos étnicos, la realización de meta-análisis que validen las asociaciones encontradas, la determinación de requisitos mínimos en la evidencia para las asociaciones genéticas y la proposición de guías clínicas consenso. Al respecto, varios centros pioneros han iniciado programas medicamentos genómicos70. Un ejemplo es el Centro Médico de la Universidad Vanderbilt, EE.UU., donde los pacientes programados para angiografía coronaria son preventivamente genotipados en una plataforma con 184 variantes, incluyendo CYP2C19. Las recomendaciones basadas en el genotipo se proporcionan automáticamente de manera electrónica para que los médicos tengan acceso cuando requieran recetar una terapia antiplaquetaria. La utilización clínica de este programa farmacogenómico ha permitido a la Universidad de Vanderbilt desarrollar soluciones a las barreras logísticas, financieras y de ejecución de conocimientos y, junto con otros programas ha generado marcos de consenso.
Una propuesta más avanzada al respecto es la reciente sugerencia de desarrollar algoritmos de dosificación, como ya se ha hecho para la warfarina, a fin de evitar el exceso de exposición a medicamentos de uso cardiovascular71. Los autores proponen considerar aspectos clínicos y demográficos (sexo, edad, índice de masa corporal, origen étnico, dosis y el tiempo de la última dosis), genéticos (variantes genéticas en CYP2C9, SLCO1B1 y ABCG2) y metabólicos (4 -hidroxicolesterol) para formular algoritmos de dosificación que definan la respuesta a rosuvastatina y atorvastatina. Los primeros resultados muestran que estos algoritmos predicen la respuesta de aproximadamente el 50% de los pacientes que toman las dosis más altas de la práctica clínica habitual, la exposición sistémica a estatina supera el percentil 9072. Sin embargo, la utilidad clínica mundial de estos algoritmos necesita ser validada.
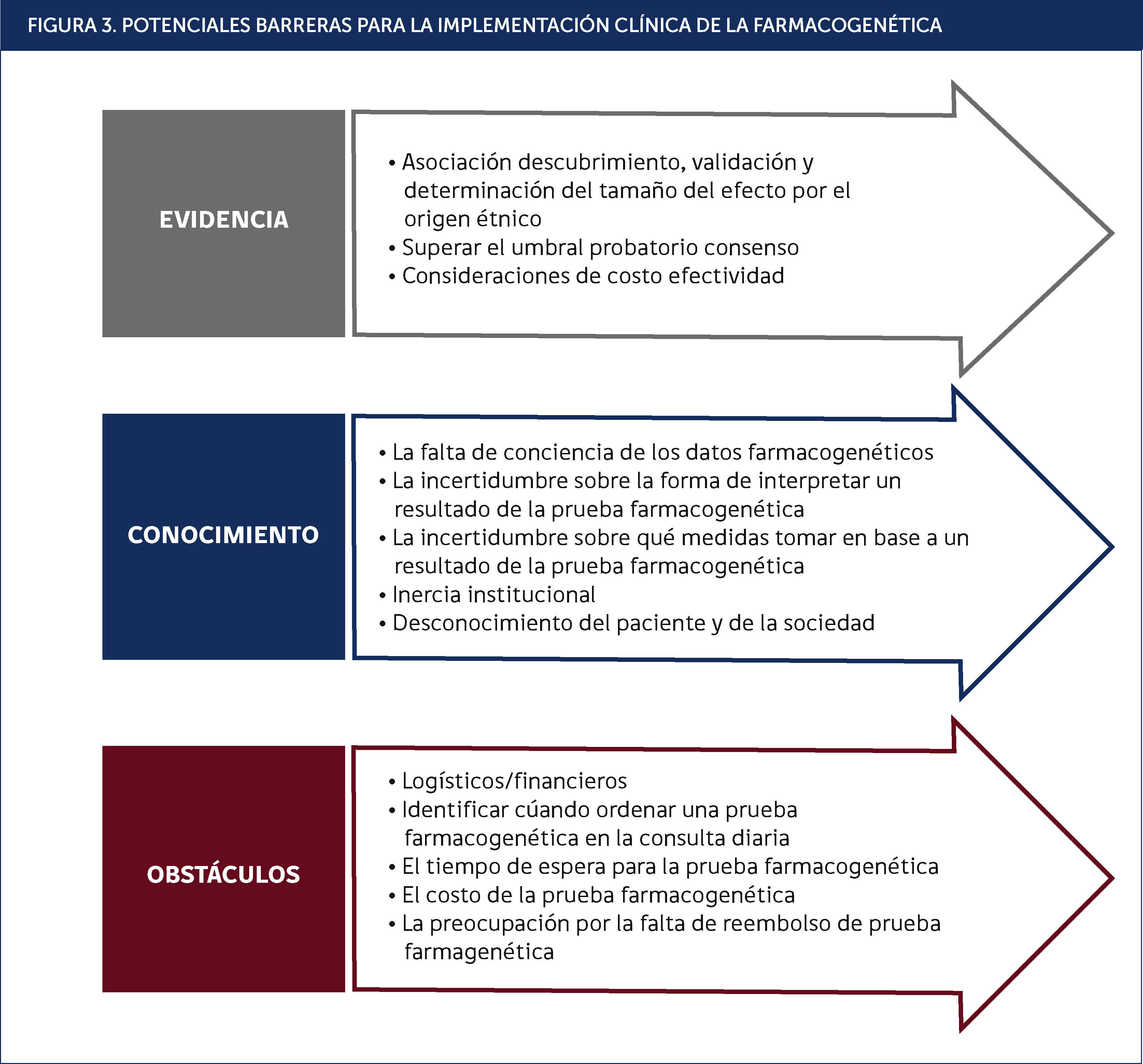
Las barreras que obstruyen la aplicación generalizada de la farmacogenómica mundialmente se pueden clasificar en limitantes de evidencia o conocimiento y oportunidad logística/financiera para su aplicación (Figura 3)21. Dentro de la evidencia en literatura científica la identificación de variantes genéticas relevantes en cada etnia y la demostración del beneficio clínico es un foco de mayor interés70. Para ilustrar esto, podemos ejemplificar el camino que ha llevado a los genes BRCA a ser una prueba genética preventiva de alta credibilidad que conduce incluso a decisiones de intervención radical (ej. mastectomía profiláctica), pero ello requirió una amplia y sustancial base de pruebas científicas y clínicas antes de la implementación.

Adicionalmente, existe una necesidad de homologar los criterios utilizados para la aceptabilidad de las pruebas genéticas y no genéticas; por ejemplo, actualmente aceptamos un cambio en la dosis de algunos medicamentos cuando se administra a pacientes con insuficiencia renal en base a los datos obtenidos a través de los estudios farmacocinéticos, pero no se acepta el mismo tipo de datos cuando un fármaco se administra a un paciente con una variante (polimorfismo) genética en una vía metabólica, cuando el cambio en la exposición sistémica es equivalente21.
Los obstáculos logísticos o financieros incluyen la falta o exceso de solicitud de análisis de los médicos, los tiempos de respuesta de las pruebas, el costo de la pruebas farmacogenéticas y la preocupación por la falta de reembolso en los sistemas de salud21.
Existen además importantes barreras del conocimiento tanto por parte del profesional clínico como del paciente. El desconocimiento de la posibilidad contar con pruebas farmacogenómicas o la falta de familiarización con la base actual de evidencias moleculares e interpretación de pruebas farmacogenómicas resulta una limitante importante. Esta brecha de conocimiento podría abordarse a través de programas educativos institucionales o nacionales dispuestos para la comunidad biomédica y los pacientes. Del mismo modo, el considerar los aspectos éticos en los procedimientos de análisis farmacogenómicos debe incluirse como una obligatoriedad en la relación médico-paciente para estos efectos.
Mirando hacia el futuro, es evidente la necesidad de una mejor orientación sobre cómo biomarcadores deben ser objeto de calificación, qué tipo de pruebas son aceptables y una mayor armonización entre los organismos reguladores de todo el mundo y los centros clínicos. Esto proporcionará rutas más claras para el reembolso y la aplicación dentro de la práctica clínica. Es importante que los investigadores sean cautelosos en sus recomendaciones en la fase de descubrimiento y la reproducción de cualquier biomarcador aplicable en las diversas poblaciones de pacientes.
En Chile algunas prácticas aisladas se han realizado, respaldadas por nuestro grupo de investigación, en diversos hospitales y centros clínicos, donde se están genotipificando variantes para explicar la variación interindividual de fármacos para la depresión y el cáncer.
Por último, esta revisión se ha centrado en la variación genómica/farmacogenómica, aunque existen otras tecnologías “ómicas” también interesantes (epigenómica, transcriptómica, proteómica, metabolómica y metagenómica), con mayor distancia de aplicación clínica. Hemos centrado además nuestra exposición acerca del estado del arte del área en aquellos medicamentos de uso cardiovascular y genes relacionados que poseen mayor evidencia científica.
En conclusión, a pesar que la farmacogenómica cardiovascular está en etapas aún iniciales y los resultados en muchos casos son aún contradictorios, es nuestra expectativa que la farmacogenómica de medicamentos cardiovasculares pueda ofrecer un beneficio real para el paciente, superando eso sí los obstáculos anteriormente expuestos. Lento, pero seguro, con esmero y con arduo trabajo la farmacogenómica cardiovascular está posicionándose como una herramienta de corriente principal en la práctica clínica, aunque queda mucho por realizar.
Los autores declaran no tener conflictos de interés, en relación a este artículo.