



Entre el 20 a 40% de los tumores cerebrales pueden manifestarse primariamente con crisis epilépticas y un 20 a 45% pueden presentar epilepsia durante el curso de la enfermedad. Las crisis pueden ser causadas por el compromiso cortical tumoral, así como en áreas distantes por deaferentación. Las crisis pueden responder a fármacos antiepilépticos o presentarse como epilepsia refractaria a fármacos. Los tumores de más lento crecimiento se asocian a epilepsia de largo tiempo de evolución, pero hay un grupo especial de neoplasias (gangliogliomas y tumores disembrioplásticos neuroepiteliales), donde la epilepsia puede ser única manifestación clínica.
La Resonancia Magnética de cerebro es mandatoria en el estudio de todo paciente con epilepsia, para detectar lesiones estructurales, especialmente en epilepsia focal. Alrededor del 30% de los pacientes operados de epilepsia refractaria presentan tumores. En estos casos el control de crisis post-operatorio llega hasta un 70% en el seguimiento a largo plazo.
Seizures occurs in 20-40% of patients with brain tumors. Seizures are caused by the tumor because of involvement with the surrounding cortex or more distal areas and may respond to antiepileptic drugs or produce a chronic, intractable seizure disorder. Within tumor pathology the slow growing neoplasms are more associated with chronic epilepsy, in gangliogliomas and dysembrioplastic neuroepithelial tumors usually seizure is the only symptom.
The MRI is mandatory in epilepsy in order to find structural lesions especially in focal epilepsy. Around 30% of all patients with surgical treatment of refractory epilepsy have neoplastic lesions as histopathological findings and the seizure control in this group is more than 70% in the long follow up.
Las crisis epilépticas pueden ser en el 20 a 40% de los pacientes, la primera o única expresión clínica de un tumor intracraneano, exceptuando los tumores de hipófisis y fosa posterior1,2. Por otra parte, un 20 a 45% de los casos pueden presentar epilepsia durante el curso de la enfermedad. La epilepsia afecta significativamente la calidad de vida de los pacientes, especialmente si esta persiste pese a uso de fármacos3,4.
Del total de pacientes portadores de epilepsia, cerca de un 6% es secundaria a una lesión neoplásica (LN). El principal grupo etario que debuta con crisis epilépticas y su causa es una LN son los adultos en la edad media de la vida (30%)1,2,5.
La incidencia de crisis se relaciona con la histología de la lesión y su localización en la corteza cerebral1–3. Las neoplasias de lento crecimiento son más epileptógenas, así en orden decreciente se sitúan los tumores glioneuronales (Ej.: ganglioglioma y tumor disembrioplástico neuroepitelial), gliomas de bajo grado, oligodendrogliomas, meningiomas y glioblastomas multiformes. Sin embargo, algunas LN parecen tener características diferentes en los pacientes portadores de epilepsia fármaco resistente crónica (EFRC). Ya que en este grupo de pacientes los gliomas de bajo grado cursan generalmente con una larga historia de epilepsia como único síntoma y post cirugía suelen no presentar recidivas tumorales1,3,5,6.
El “Registro de Europeo de Neuropatología de Cirugía de Epilepsia”, con sede en la Universidad de Erlangen (Alemania), reporta un total de 5842 pacientes operados de epilepsia refractaria. De ellos el 26.5% correspondía a neoplasias, la edad promedio al momento de la cirugía fue de 18.5 años y la duración promedio de su epilepsia de 12.5 años. Es decir, un largo tiempo con crisis epilépticas como único síntoma7, de allí lo importante de realizar estudio con Resonancia Magnética (RM) de cerebro en todo paciente sospechoso de presentar crisis epiléptica focal o que no presente un síndrome epiléptico benigno8,9.
MECANISMOS CAUSALES DE EPILEPSIAEl mecanismo celular intrínseco que provoca las crisis epilépticas permanece aún incierto. Sin embargo, existen varias hipótesis de como las LN pueden llegar a producir epilepsia: compromiso tumoral de la corteza sana con alteración del ácido gamma amino butírico (GABA) intra cortical, producción de impulsos citotóxicos, deaferentación y degeneración transináptica a distancia5,10. Estas últimas hipótesis se ven avaladas por la mayor asociación de esclerosis del hipocampo en pacientes con lesiones temporales, lo anterior también se denomina patología dual (esclerosis del hipocampo + otra lesión)5,10–16.
Además generalmente las LN, tales como gangliogliomas (GG) y tumores disembrioplásticos neuroepiteliales (DNET o DNT) suelen presentar un foco epileptógeno en la corteza perilesional, donde suele existir displasia cortical focal asociada (DCF) a la neoplasia5–7. Las DCF son altamente epileptógenas y estas corresponden a la primera causa de cirugía de la epilepsia en niños14. Los tumores asociados a DCF pertenecen al tipo IIIb de la clasificación de displasias de la Liga Internacional contra la Epilepsia11.
La literatura ha sugerido que la lesión misma pudiera ser la causante de la epilepsia debido a la producción de neurotransmisores excitatorios. Es decir, todas las hipótesis postuladas indican que la lesión neoplásica es previa al inicio de la epilepsia. Esto cobra aún mayor importancia al analizar el largo tiempo de epilepsia como único síntoma (generalmente mayor a 10 años) en todas las series quirúrgicas de EFRC6,7,12–16, lo cual indicaría que la mayoría de los tumores han permanecido largo tiempo en el encéfalo y han crecido muy lentamente o bien han permanecido estáticos, siendo desconocido el real tiempo de permanencia en el parénquima cerebral; incluso es posible plantear, especialmente en GG y DNET, que se traten de lesiones congénitas instaladas durante la formación de la corteza cerebral11,17.
Además este tipo de LN no muestran los hallazgos típicos observados en gliomas infiltrantes, tales como mutaciones de IDH1 o deleciones de 1p/19q. En contraste marcadores onco-fetales como la proteína CD34 puede ser frecuentemente identificada y existen genes del desarrollo comprometidos. Mutaciones en la proteína B-RAF o de la proteína “blanco de rapamicina en células de mamífero” (mTOR) han sido también identificadas como hallazgo en este tipo de tumores7.
Otras causas de crisis epilépticas asociadas a tumores pueden ser a causa de su tratamiento mismo, especialmente en tumores de alto grado, como por la cirugía misma (inflamación con edema, hemorragia, gliosis, etc.), quimioterapia (especialmente con el uso de vincristina, L-asparginasa o ciclosporina) y radioterapia (vasculopatía)5.
TRATAMIENTOA) Tratamiento médico con fármacos antiepilépticos (FAEs): Es mandatorio en pacientes con epilepsia. Las crisis en LN suelen ser focales, donde la semiología de la crisis dependerá del lóbulo afectado y su propagación, o bien crisis focales secundariamente generalizadas1–3. Por esto la recomendación es utilizar FAEs de primera línea en monoterapia para crisis focales, las sugerencias varían según las diferentes guías clínicas, pero tienden a recomendar como primera línea a: carbamazepina (idealmente de liberación prolongada), levetiracetam, lamotrigina o fenitoina18,19. Se debe tener mucha precaución en identificar eventual efectos secundarios de los FAEs, que pueden dañar la calidad de vida del paciente, tales como: sedación, interferencia cognitiva, alteraciones psiquiátricas, interacción con otros fármacos, etc.2,18,19.
Nuevos FAEs como lacosamida pudieran ser también una alternativa por no presentar interacción con la mayoría de otros fármacos20.
El uso de FAEs profilácticos en pacientes con tumores cerebrales y sin crisis no está recomendado por la academia americana de neurología, pero en la practica médica habitual si se utiliza. Sin embargo, pese al uso de un FAE bien seleccionado, el éxito de libertad de crisis no supera el 50% con el uso de un FAE de primera línea y un 13% adicional con un segundo FAE de primera línea, aproximadamente un tercio de los pacientes persisten con crisis, pese a usar FAEs adecuados en mono o politerapía21.
B) El tratamiento quirúrgico: Puede ser “lesional” si el paciente no presenta EFRC o existe hipertensión endocraneana. La cirugía debe ser idealmente con la resección completa de la lesión, a menos que esté ubicada en corteza elocuente y el paciente no acepte un déficit neurológico definitivo. Sin embargo, algunos casos de lesiones de bajo grado de larga evolución pueden tener desplazada el área funcional clásica por neuroplasticidad22–24.
En pacientes con EFRC, la recomendación es hacer un abordaje “funcional” y realizar un estudio pre-quirúrgico con a lo menos video-EEG y evaluación neuropsicológica. Además algunos casos seleccionados pueden requerir estudios adicionales de RM funcional, PET, electrocorticografía intraoperatoria (EcoG), estudios invasivos, entre otros.8–10,25–27.
HISTOLOGÍA EN TUMORES Y EPILEPSIA REFRACTARIALa cirugía de la epilepsia refractaria a fármacos se asocia en alrededor de un 30% a tumores de bajo grado, pero destaca que sobre el 60% de ellos corresponden a gangliogliomas y tumores disembrioplásticos neuroepiteliales7. Sin embargo, estos 2 tipos de tumores no superan el 1% de las neoplasias en la práctica neuroquirúrgica habitual6,7,13. Estos tumores presentan frecuentemente una larga historia de epilepsia como único síntoma y su excelente evolución post-operatoria, esto indica que presentan una conducta biológica diferente a lo conocido previamente para gliomas y tumores de bajo grado en general25. Incluso algunos autores han propuesto el término de “epileptomas” a estas lesiones, para ayudar a un mejor análisis entre epileptólogos, neuropatólogos y neurooncólogos7.
Entonces, se debe tener mucho cuidado en aplicar un criterio de cirugía oncológica en estos casos, ya que no solo se debe resecar la lesión, sino, también el foco epileptógeno, el cual no necesariamente corresponde solo a la lesión, sino que puede ubicarse en su periferia o incluso a distancia por deaferentación crónica.
A continuación se revisan los 2 tipos de tumores más frecuentemente asociados a epilepsia crónica.
GANGLIOGLIOMA (GG)Historia e incidencia: El término GG fue acuñado por Perkins en 192628. Es un tumor infrecuente del sistema nervioso central (SNC), con una incidencia de entre el 0.4 a 1.3% en adultos. Es más frecuente en niños, donde alcanza una frecuencia de hasta un 9%. No existe diferencia de preponderancia según los distintos sexos29–34.
Histopatología: El GG está constituido por células ganglionares maduras atípicas, situadas en una matriz glial frecuentemente astrocitaria y en algunos casos, también con oligodendrocitos35,36. El tumor es considerado una combinación neuro-glial, según la Organización Mundial de la Salud (OMS). El GG histopatológicamente puede ser confundido con un hamartoma o con DCF, debido a que ambas lesiones están compuestas también por glías y elementos neuronales, e incluso displasias corticales pueden ser lesiones acompañantes de un GG en hasta un 20% de los casos11,13. Esto es aún más relevante, cuando se trata de lesiones pequeñas o bien la muestra operatoria es escasa. El GG, también puede ser confundido con un astrocitoma, ya que las células ganglionares pueden ser escasas primando el componente astrocitario. Entonces el patólogo puede pensar que se trata de células ganglionares envueltas, por el crecimiento de un astrocitoma. Lo anterior se soluciona usando los criterios de Russell y Rubinstein, para el correcto diagnóstico histopatológico:
- A)
El GG está compuesto por una mezcla de células gliales y neuronas.
- B)
Las células gliales son frecuentemente astrocitos.
- C)
Las células son identificadas como neuronas, solo si la sustancia de Nissl puede ser demostrada por tinción de cristal violeta o si ellas dan origen a procesos neuronales demostrados por tinción de cuerpos modificada de Bielschowski; para que las neuronas sean catalogadas de neoplásicas, ellas deben ser claramente heterotópicas (localizadas lejos de la sustancia gris) o atípicas mostrando desorientación, formas y tamaños bizarros, núcleos con hipercromatismo y frecuentemente binucleación36.
El uso de tinciones inmuno-histoquímicas, ya sea para astrocitos (proteína ácida glio-fibrillar {GFAP}) o para neuronas (sinaptofisina, proteína de neurofilamento {NFP} y enolasa especifica para neuronas {NSE}); ayudan al diagnóstico de GG, sobre todo en el caso de tumores pequeños o muy fragmentados. Es probable que estas consideraciones no hayan sido hechas en publicaciones previas, lo cual ayudaría a la baja incidencia de GG, al ser confundido con astrocitomas o hamartomas. Además es útil poder usar otros marcadores inmuno-histoquímicos, tales como anticuerpos CD34 y MAP27.
Localización: Su ubicación puede ser en cualquier parte del SNC: lóbulos cerebrales29–34, cordón espinal37, unión bulboespinal38, tronco cerebral, cerebelo39, región pineal, tálamo o nervio óptico, pero es más frecuente, en la región supratentorial, con preferencia en el lóbulo temporal6,7,29–32.
Manifestación clínica: La mayoría de los pacientes presentan una larga historia de epilepsia parcial compleja como síntoma único6,7,15. Esto dado que la principal ubicación del tumor es el lóbulo temporal7,13,30. También se puede asociar a distintos trastornos psiquiátricos crónicos, incluida la esquizofrenia40.
Los déficit neurológicos focales dependen de la ubicación del tumor. En la región infratentorial estos son raros de encontrar, así como el aumento de presión intracraneana. Esto debido a que es un tumor de lento desarrollo e incluso se puede plantear que muchos casos se tratarían de lesiones estáticas29–32.
Anaplasia: Dada su baja incidencia se sabe poco sobre su historia natural y conducta biológica. El GG se considera más bien un tumor de baja malignidad, de lento crecimiento, no infiltrante, su malignización es rara y cuando esta ocurre es a partir del componente glial, especialmente de la parte astrocítica41. No existe actualmente guías clínicas que orienten al grado de recurrencia o malignización7.
Neuroradiología- 1
- Tomografía axial computarizada (TAC): Tiene un bajo rendimiento en la detección de GG pequeños, sólo supera a la RM en la detección de pequeños focos de calcificación en la masa tumoral, que pueden pasar desapercibidos en la RM. El GG suele ser hipodenso entre un 40 a 100%, con calcificaciones entre un 20 a 50% y tomar el medio de contraste entre un 16 a 80%30,34,42,43.
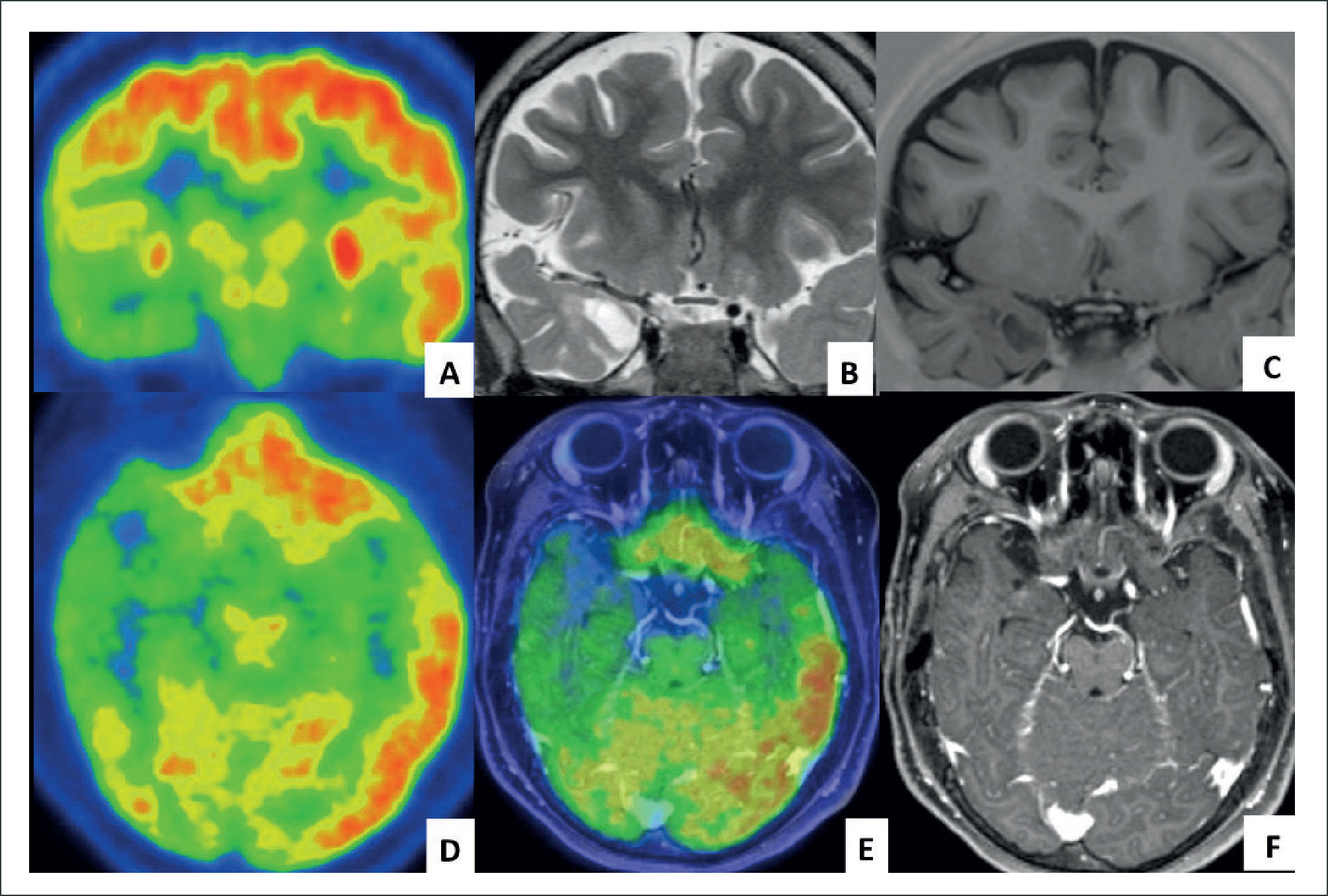
 PET (tomografía por emisión de positrones), corte coronal, con hipo-metabolismo temporal mesial y lateral derecho (color verde), B) RM fase T2 coronal, con lesión quística temporal mesial delante de la amígdala, con alteración de la sustancia blanca. C) RM fase T1-IR con lesión sin edema, ni efecto expansivo. D) PET axial con extenso hipometabolismo temporal lateral derecho. E) Fusión de RM volumétrica T1 con contraste y PET, donde se hace más evidente el área alterada funcionalmente. F) RM fase T1 con gadolinio, donde no se ve impregnación anormal.") FIGURA 1.
FIGURA 1.IMÁGENES DE PACIENTE DE 9 AÑOS, OPERADO AL AÑO DE VIDA DE GANGLIOMA, CON RESECCIÓN SUB-TOTAL
Consulta por epilepsia refractaria y severo deterioro cognitivo. Las imágenes muestran: A) PET (tomografía por emisión de positrones), corte coronal, con hipo-metabolismo temporal mesial y lateral derecho (color verde), B) RM fase T2 coronal, con lesión quística temporal mesial delante de la amígdala, con alteración de la sustancia blanca. C) RM fase T1-IR con lesión sin edema, ni efecto expansivo. D) PET axial con extenso hipometabolismo temporal lateral derecho. E) Fusión de RM volumétrica T1 con contraste y PET, donde se hace más evidente el área alterada funcionalmente. F) RM fase T1 con gadolinio, donde no se ve impregnación anormal.
(0.38MB). - 2
- Resonancia magnética: No ofrece un patrón clásico30,34,42,43. Se suele encontrar: A) Falta de efecto expansivo sobre el parénquima cerebral peri-tumoral, incluso con amplias cisternas de la base en tumores temporo-mediales. Estos son signos que indicarían una larga permanencia del tumor. B) Ausencia de edema peri-lesional. C) Presencia de quiste, generalmente único (Figura 2). Dado lo anterior, las imágenes en la RM son generalmente: A) una señal hipointensa en T1 e hiperintensa en T2 producto de la lesión quística. B) Las lesiones sólidas aparecen homogéneamente hiperintensa en la fase T2, pudiendo incluso aparecer isointensas en T1. Todos los puntos anteriores son sólo una orientación, dada la alta similitud en la RM de GG con astrocitomas de bajo grado. Sólo la histopatología certifica el correcto diagnóstico.
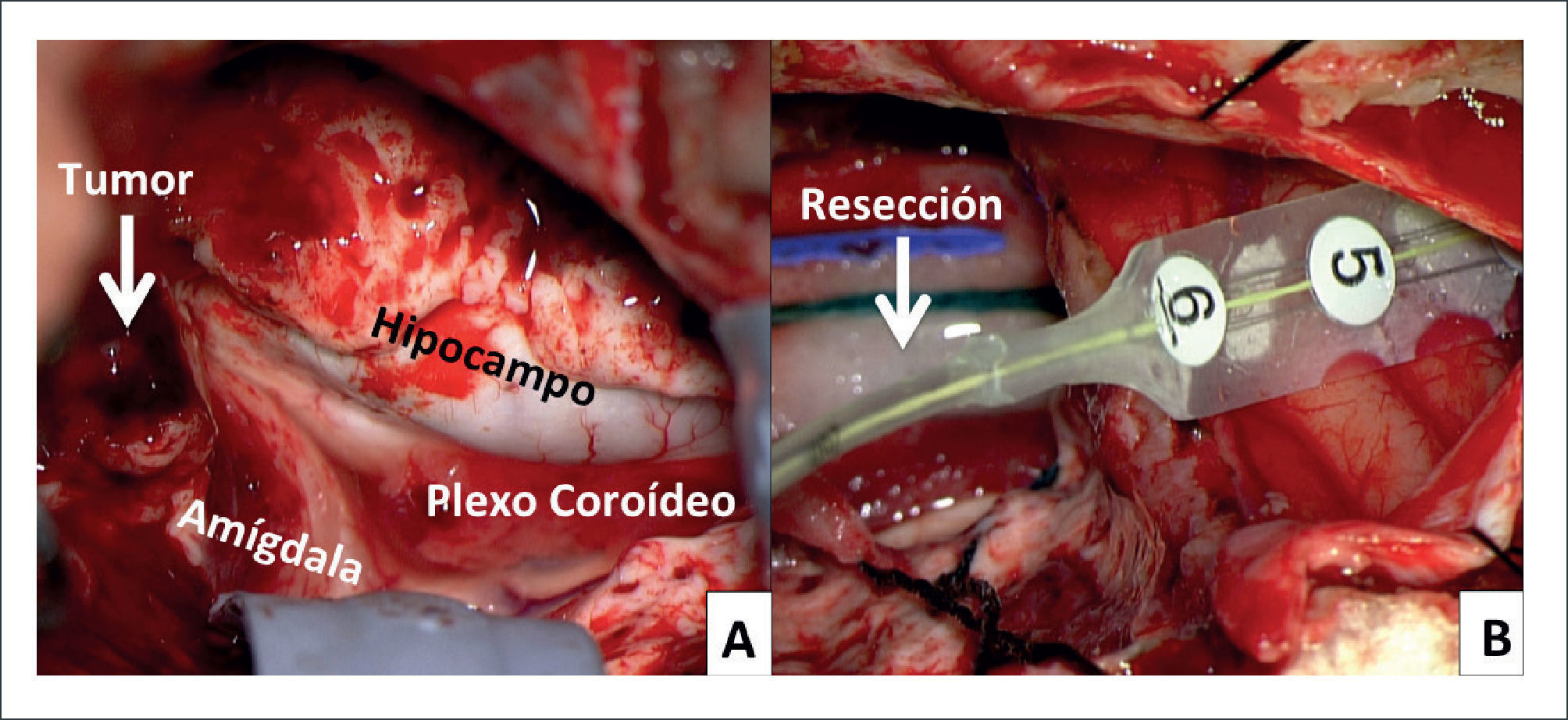
 Post-resección de neocorteza del polo temporal derecho. Tumor (flecha) en porción anterior de la amígdala e hipocampo. B) Electrocorticografía final en corteza temporal lateral posterior, post resección de ganglioglioma + tejido epileptógeno (amígdala e hipocampo).") FIGURA 2.
FIGURA 2.FOTOS INTRA OPERATORIAS DE PACIENTE DE FIGURA 1
A) Post-resección de neocorteza del polo temporal derecho. Tumor (flecha) en porción anterior de la amígdala e hipocampo. B) Electrocorticografía final en corteza temporal lateral posterior, post resección de ganglioglioma + tejido epileptógeno (amígdala e hipocampo).
(0.31MB). - 3
-Tomografía por emisión de positrones (PET-CT): Este examen se puede realizar inter-ictal (entre crisis) o peri-ictal (muy cercano a las crisis), siendo el peri-ictal el más frecuente. Los hallazgos pueden mostrar hipo-metabolismo en la zona funcionalmente comprometida o hiper-metabolismo en la zona tumoral. En nuestra institución realizamos la inyección del radio-fármaco (FDG-glucosa marcada), mientras el paciente está video-EEG en una habitación al lado del equipo de PET-CT, de modo de conocer su actividad eléctrica al momento de examen; ya que sea el paciente está con crisis clínicas o sub-clínicas se puede incluso encontrar hiper-metabolismo en la zona epileptógena.
 PET (tomografía por emisión de positrones), corte coronal, con hipo-metabolismo temporal mesial y lateral derecho (color verde), B) RM fase T2 coronal, con lesión quística temporal mesial delante de la amígdala, con alteración de la sustancia blanca. C) RM fase T1-IR con lesión sin edema, ni efecto expansivo. D) PET axial con extenso hipometabolismo temporal lateral derecho. E) Fusión de RM volumétrica T1 con contraste y PET, donde se hace más evidente el área alterada funcionalmente. F) RM fase T1 con gadolinio, donde no se ve impregnación anormal.")
 Post-resección de neocorteza del polo temporal derecho. Tumor (flecha) en porción anterior de la amígdala e hipocampo. B) Electrocorticografía final en corteza temporal lateral posterior, post resección de ganglioglioma + tejido epileptógeno (amígdala e hipocampo).")
Tratamiento: La resección tumoral completa es el tratamiento de elección, el cual es considerado curativo en los casos de GG de bajo grado de malignidad29–34. Dado que muchos pacientes presentan EFRC, se debe hacer en estos casos, una completa evaluación funcional prequirúrgica, para la localización del foco epileptógeno, ya que como se mencionó, la lesión cerebral no es sinónimo de foco epileptógeno. Entonces en muchos casos la sola resección del tumor no bastará para controlar las crisis epilépticas44,45. Nosotros recomendamos el uso de electrocorticografía (EcoG) intra operatoria, para determinar mejor el área irritativa, los estudios invasivos solo en casos seleccionados (Figura 2).
El control de las crisis fluctúa entre el 50-90%. Los resultados son mejores en los centros, que realizan cirugía funcional, es decir, la localización pre-operatoria del foco epileptógeno y su resección junto al tumor8,9,29–34.
La radioterapia hasta ahora no ha mostrado utilidad. Dado los pocos casos de anaplasia, el beneficio de la radioterapia o quimioterapia no ha sido comprobado41.
TUMOR DISEMBRIOPLÁSTICOS NEUROEPITELIALES (DNET)Historia: El DNET es una lesión muy infrecuente del SNC. Este fue descrito por primera vez en el año 1988 por la neuropatóloga Daumas-Duport46 e incluido el 1993 en la clasificación de tumores cerebrales de la OMS, dentro de la categoría de tumores neuronales y por combinación neuroglial.
El DNET se caracteriza por ser una lesión asociada frecuentemente a epilepsia crónica, usualmente sin otro déficit neurológico agregado. Su ubicación es generalmente supratentorial y frecuentemente localizado en el lóbulo temporal5,7,45,46.
Neuropatología: el DNET es una lesión intracortical asociada con DCF, su arquitectura es multinodular y está compuesta por astrocitos, oligodendrocitos y neuronas. Estas últimas están situadas generalmente en una matriz mixoidea7,46,47. La misma autora, 11 años después48, lo sub-clasificó en tumor DNET simple, es decir, conteniendo sólo elementos glioneuronales, y complejo, en el que a los elementos glioneuronales se añadían estructuras columnares, nódulos gliales y focos de displasia cortical. Esta especial relación entre el tumor y la displasia cortical es la causa principal del debate que aún existe sobre esta entidad, ya que algunos no lo consideran verdaderamente un tumor, sino parte del espectro de las displasias.
DNET y epilepsia: Se introdujo el término DNET luego de un estudio retrospectivo en un grupo de pacientes jóvenes con complejos tumores neuroepiteliales muy difíciles de clasificar en la lista de tumores de la OMS y asociados a epilepsia fármaco-resistente46, quienes fueron operados en el Hospital Santa Ana de París y en la Clínica Mayo de Rochester. Estos pacientes habían sido previamente clasificados generalmente como oligodrendrogliomas, pero llamaba la atención su sobrevida prolongada y su libertad de crisis epilépticas. Las series de DNET lo confirman como una lesión asociada a epilepsia, pero que no es sólo propia de pacientes jóvenes49.
En un estudio a lo largo de 10 años sobre 327 resecciones consecutivas en pacientes con epilepsia parcial, mostró al DNET, como el tumor más frecuente en EFRC50.
Localización y probable origen: Los DNET son de ubicación supratentorial. La localización es usualmente en el lóbulo temporal especialmente en el hipocampo o cerca de él. Esto se puede asociar al probable origen de los DNET, ya que el hipocampo tiene una matriz germinal que aparece en la 12° semana del desarrollo embrionario, estas células son neuro-epitelio primitivo, las cuales se expanden rápidamente y cubren completamente la convexidad de la corteza entre las 16 a 18 semanas de gestación, originando así la capa granular de la corteza. Esta capa involuciona producto de la migración de estas células dentro de la corteza cerebral, donde ellas madurarán entre las células gliales51. También han sido reportados casos de DNET con compromiso multifocal de diversos sitios del SNC52.
Cabe hacerse la pregunta: ¿Es realmente el DNET un tumor (neoplasia) o es una lesión malformativa del desarrollo cortical? Los argumentos a favor de una lesión malformativa son:
- 1)
Se han comunicado escasos casos de DNET con anaplasia, recurrencia o malignización luego de una resección53.
- 2)
La hipótesis del origen embrionario del DNET permite relacionar esta lesión a una malformación del desarrollo (MDC) más que a un tumor propiamente tal. Puede ser que el DNET, así como el GG y las displasias corticales, sean sólo diferentes grados de manifestación de una MDC durante la embrio- génesis10,54.
Histología: Histológicamente el DNET presenta múltiples nódulos de diferentes tamaños (0.5-3mm)47,54, a menudo rodeando la corteza extra-nodular el tumor presenta el llamado “componente especifico”, el cual consiste en una gran cantidad de oligodendrocitos y cambios micro-quísticos con acumulación de una matriz musinosa entre las células, donde las neuronas parecen “flotar”51. Esta última característica puede ser la responsable del aspecto poliquístico del DNET en la RM. Los tumores de la serie original de Daumas-Duport fueron previamente clasificados como: oligodendrogliomas, astrocitomas u oligoastrocitomas46.
Neuroradiología:- 1
- TAC: Las imágenes más características muestran una lesión hipodensa, con variable aspecto pseudo-quístico, alteración de la tabla interna del hueso supra-yacente en 1/3 de los casos, como signo de larga evolución55.
- 2
- RM: El DNET se presenta generalmente como una lesión poli-microquística que da una imagen tipo “panal de abejas” (Figura 3). El DNET tiene baja intensidad de señal en T1 y señal hiperintensa en T247,51,55,56. La matriz musinosa del DNET da en general una intensidad de señal más baja al LCR en T1, pero no difiere del LCR en T2. En FLAIR se suele ver hiperseñal en la lesión. El componente sólido del DNET puede presentar captación del medio de contraste como otros tumores.
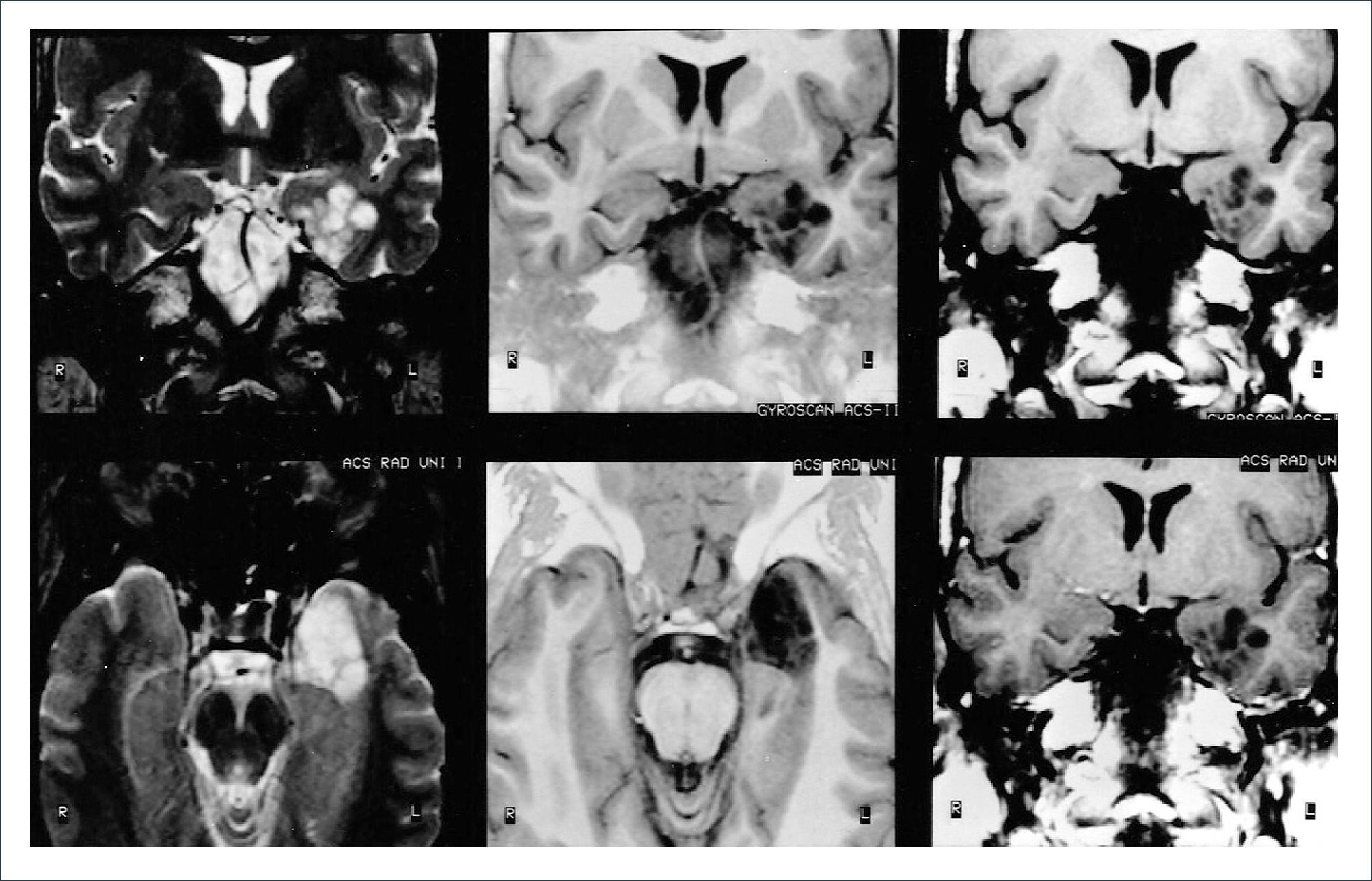
, al centro T1-IR (coronal arriba, axial abajo) y a derecha T1 coronales. Todas las imágenes muestran un tumor temporal mesial izquierdo, con lesiones poli-micro-quísticas, tipo panal de abejas, sin edema, ni efecto expansivo. Estos son los hallazgos típicos de un DNET.") FIGURA 3.
FIGURA 3.RM EN FASES
T2 a izquierda (coronal arriba, axial abajo), al centro T1-IR (coronal arriba, axial abajo) y a derecha T1 coronales. Todas las imágenes muestran un tumor temporal mesial izquierdo, con lesiones poli-micro-quísticas, tipo panal de abejas, sin edema, ni efecto expansivo. Estos son los hallazgos típicos de un DNET.
(0.35MB). - 3
- PET-CT: Presenta las mismas características que en GG, pero dadas las lesiones poli-quísticas es más frecuente de encontrar hipo-metabolismo en la zona tumoral.
, al centro T1-IR (coronal arriba, axial abajo) y a derecha T1 coronales. Todas las imágenes muestran un tumor temporal mesial izquierdo, con lesiones poli-micro-quísticas, tipo panal de abejas, sin edema, ni efecto expansivo. Estos son los hallazgos típicos de un DNET.")
Tratamiento quirúrgico: La cirugía debe realizarse resecando completamente la lesión y el tejido epileptogénico perilesional, intentando no lesionar zonas elocuentes5,8. En cirugía de la epilepsia temporal las resecciones pueden ser estándar o resecciones a la medida del paciente6,10,15,22, esta última parece ser la más aconsejable.
Las neuroimágenes de control muestran tumor residual hasta en el 20% de los pacientes, pero con control satisfactorio de las crisis30,48,57.
SÍNTESISLas neoplasias cerebrales suelen presentarse con crisis epilépticas hasta en el 40% de los casos, generalmente con crisis focales y su semiología depende de su ubicación. La imagen de elección es la RM de cerebro en todo paciente que inicia crisis epilépticas especialmente en adultos jóvenes. Los tumores de bajo grado se asocian a epilepsia de largo tiempo de evolución. El GG y el DNET son las lesiones más frecuentemente asociadas a epilepsia refractaria y se encuentran en la frontera entre lesiones malformativas y tumores propiamente tales. La cirugía puede ser lesional en casos de reciente aparición de la epilepsia o funcional (identificación del foco epileptógeno), en casos de epilepsia refractaria. En general los tumores asociados a epilepsia crónica presentan una excelente conducta biológica (baja morbi mortalidad) y un buen control de crisis post-operatoria, llegando a un promedio de un 70% de libertad de crisis en el largo tiempo.
El autor declara no tener conflictos de interés, en relación a este artículo.