





P-246 - PECOMA EN PACIENTE JOVEN CON RECIDIVA TUMORAL DE ORIGEN MESENQUIMAL. UN GRUPO DESCONOCIDO DE TUMORES
Hospital Universitario de La Princesa, Servicio de Cirugía General y del Aparato Digestivo, Hospital Universitario de La Princesa, Instituto de Investigación Sanitaria Princesa (IIS-IP), Universidad Autónoma de Madrid (UAM), Madrid.
Introducción: El término PEComa engloba un grupo heterogéneo e infrecuente de tumores mesenquimales, siendo alguno de ellos el angiomiolipoma, la linfangioleiomatosis o el tumor de células claras tipo sugar extrapulmonar. Estos tumores pueden asociarse con trastornos genéticos como la esclerosis tuberosa (ET) o el reordenamiento del factor de transcripción TFE3. Debido a su infrecuencia, la diversidad en su presentación y el tipo de paciente afecto, la sospecha diagnóstica es baja. El diagnóstico es fundamentalmente histológico, caracterizado por la presencia de cordones de vasos sanguíneos con paredes delgadas rodeados de células epitelioides; así como la positividad para marcadores melanocíticos y de músculo liso. Algunos de ellos se consideran potencialmente malignos al presentar necrosis, infiltración vascular, alto índice mitótico o gran tamaño. Presentamos el caso de una paciente joven intervenida en nuestro centro por sospecha de hepatocarcinoma fibrolamelar gigante con posterior diagnóstico anatomopatológico de PEComa.
Caso clínico: Mujer de 33 años intervenida previamente en otro centro de nefrectomía izquierda por angiomiolipoma en 2011 y bisegmentectomía (segmentos V y VI) por adenoma hepático en 2018. En el contexto de una crisis renoureteral, se diagnosticó de forma incidental mediante tomografía computarizada (TC) abdominal de una LOE en lóbulo hepático izquierdo de 10 cm de diámetro, sospechosa de hepatocarcinoma fibrolamelar. En el estudio preoperatorio destacó la negatividad de marcadores tumorales. La paciente se intervino de forma programada mediante bisegmentectomía II-III laparoscópica sin incidencias. El estudio anatomopatológico identificó un tumor mesenquimal sugestivo de PEComa con signos de malignidad dado su tamaño superior a 5 cm, más de 1 mitosis/10 cga y la presencia de necrosis e invasión linfovascular. Debido a la infrecuencia de la entidad y los antecedentes de la paciente, se consultaron las biopsias de sus cirugías previas, y finalmente se obtuvo como diagnóstico definitivo angiomiolipoma epitelioide multicéntrico renal izquierdo y angiomiolipoma epitelioide hepático; por lo que se considera la posibilidad de que la última lesión hepática no se trate de un tumor primario, sino de una metástasis de la neoplasia renal. Ante el diagnóstico final, la paciente se derivó al Servicio de Dermatología y Genética para descartar ET. La exploración dermatológica no mostró lesiones compatibles y, actualmente, está pendiente de obtener los resultados del estudio genético.
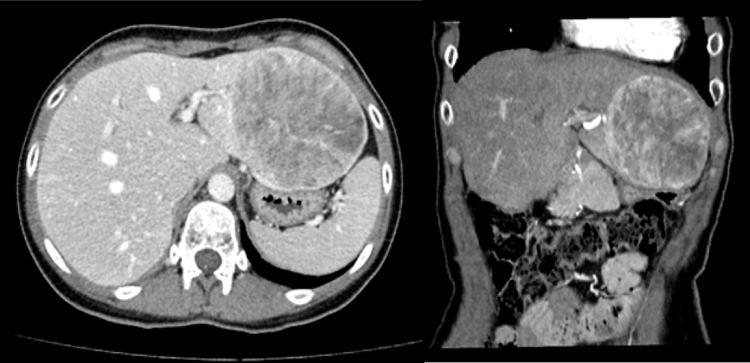
Discusión: La familia de tumores PEComa agrupan entidades de carácter benigno y maligno. Es necesario un seguimiento estrecho ante el diagnóstico de alguna de las neoplasias incluidas bajo este término (como el angiomiolipoma) especialmente cuando cumplen criterios de malignidad. Sin embargo, debido a su carácter infrecuente, no existen guías ni evidencia clara sobre cómo y cuándo realizar dicho seguimiento. No obstante, actualmente se recomienda el despistaje de síndromes genéticos que pueden asociarse a estos tumores.

