





P-622 - LEIOMIOSARCOMA DE INTESTINO DELGADO: UNA ENTIDAD INFRECUENTE
Hospital Universitario de Burgos, Burgos.
Introducción: Se presenta el caso de una mujer de 76 años, presentada en el comité de tumores por tumoración de intestino delgado de diagnóstico incierto.
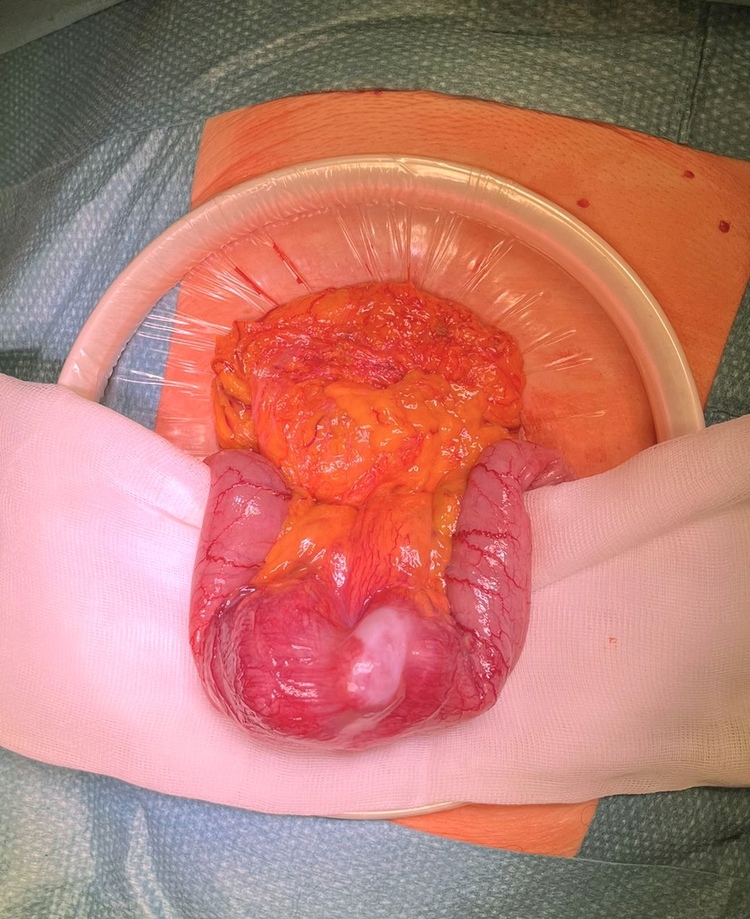
Caso clínico: Mujer de 76 años que es derivada a Consultas de Diagnóstico Rápido de Medicina Interna por anemia ferropénica (Hb 10,6). La paciente no refiere síndrome constitucional ni episodios de obstrucción intestinal. No síntomas compatibles con síndrome serotoninérgico ni otra clínica asociada. El abdomen es blando y depresible, sin masas palpables. Presenta una cicatriz infraumbilical por una cesárea previa. Se realizaron inicialmente gastroscopia y colonoscopia, sin hallazgos patológicos relevantes. Posteriormente se realiza TAC body, en el que se describe un probable engrosamiento mamelonado de asa intestino delgado a nivel pelviano, compatible con linfoma, adenocarcinoma, o GIST. Además, la paciente presenta pequeños nódulos pulmonares bilaterales, indeterminados debido al tamaño, sin poder descartar la posibilidad de metástasis. Finalmente, se realiza estudio mediante cápsula endoscópica, que en la porción media intestinal observa una estenosis de la luz, con aspecto neoplásico, que condiciona retención de la capsula. La paciente es presentada en el comité de tumores y se decide realizar cirugía para exéresis de la lesión. Se objetiva una tumoración blanquecina e indurada dependiente del yeyuno. La superficie hepática no es patológica. Se realiza una resección de intestino con márgenes laterales e incluyendo el mesenterio correspondiente, en el que se palpa una adenopatía, y se anastomosa con GIA mecánica. La anatomía patológica de la muestra es compatible con un leiomiosarcoma bien diferenciado de 7,5 × 2,5 cm, pT1, con doce ganglios con hiperplasia folicular reactiva. Posteriormente es valorada por Oncología, sin precisar tratamiento por su parte. Un año después permanece asintomática y libre de enfermedad.
Discusión: Los tumores malignos de intestino delgado suponen solamente el 2% de los tumores malignos de origen gastrointestinal. El leiomiosarcoma de ID es aún más raro, siendo el 0,13% de esos tumores. Los leiomiosarcomas son tumores raros, procedentes de células de músculo liso, que aparece sobre todo en el espacio retroperitoneal, pared vascular o tejidos blandos en las extremidades inferiores. Es un tumor excepcionalmente raro en el tracto gastrointestinal. La mayoría de los casos son esporádicos, aunque hay algunos factores de riesgo como el antecedente de retinoblastoma, VIH, pacientes inmunocomprometidos tras trasplantes o niños con inmunodeficiencias congénitas. Generalmente, el pronóstico de los sarcomas de partes blandas tiene una mediana de supervivencia de 12 meses. Se pueden presentar como masas asintomáticas, anemia por pérdidas crónicas o agudas, o como un abdomen agudo secundario a la perforación del tumor, a una invaginación o a isquemia intestinal. La base del tratamiento es la resección quirúrgica. Son tumores resistentes a la radioterapia. Dependiendo del grado histológico y el TNM, en muchas ocasiones se asocia a quimioterapia adyuvante, sobre todo teniendo en cuenta que suelen ser tumores silentes que se diagnostican en estadios avanzados.

